O gene RYR1, descrito em 1989 por MacLennan é responsável por dar as instruções para a proteína do receptor de rianodina 1 (RYR1), que é um canal de cálcio crucial para a contração e relaxamento dos músculos esqueléticos.1 A função principal do gene é codificar o canal que libera íons de cálcio do retículo sarcoplasmático (um reservatório dentro da célula muscular) quando o músculo é ativado por um sinal elétrico. Quando o canal RYR1 se abre, o cálcio entra no citoplasma, permitindo que os músculos se contraiam para permitir o movimento. Variantes (mutações) neste gene podem levar a doenças musculares como miopatias congênitas: Doença do Núcleo Central (Central Core), Doença Multi-Minicore, Miopatia Centronuclear e Desproporção Congênita do Tipo de Fibra. O estudo destas doenças permitiu o sequenciamento do gene RYR1 e a descoberta da relação de variantes neste gene e manifestações clínicas como Hipertermia Maligna (HM), Rabdomiólise e Rabdomiólise por Esforço (RE).
RYR1 está localizado no cromossomo 19q13.1 e codifica o Receptor de Rianodina Tipo 1 (RYR1). O gene RYR1 compreende 106 exons e codifica uma grande proteína com 5038 aminoácidos. Devido ao seu tamanho e complexidade consideráveis, o RYR1 tem sido historicamente um gene desafiador para estudo. Durante muitos anos, os esforços de pesquisa concentraram-se principalmente em três domínios (N-terminal, central e C-terminal) considerados pontos críticos de mutação. No entanto, com o advento das tecnologias de Sequenciamento de Nova Geração (NGS), tornou-se possível analisar o gene inteiro. Como resultado, inúmeras novas variantes foram identificadas fora dos pontos críticos previamente conhecidos. Até o momento, mais de 1000 variantes distintas do gene RYR1 foram relatadas (HGMD; https://www.hgmd.cf.ac.uk/ (acessado em 04 de dezembro de 2025) e LOVD; https://databases.lovd.nl/shared/genes/RYR1 (acessado em 04 de dezembro de 2025), porém apenas 72 foram classificadas como variantes diagnósticas pelo Grupo Europeu de Hipertermia Maligna (EMHG; https://www.emhg.org/diagnostic-mutations (acessado em 04 de dezembro de 2025)). A grande maioria dessas variantes são de sentido trocado, enquanto inserções, deleções e duplicações são relativamente incomuns, representando menos de 10% de todas as variantes patogênicas conhecidas no gene RYR1.
A Hipertermia Maligna (HM) é uma síndrome hipermetabólica farmacogenética que se manifesta como uma crise aguda após a exposição de indivíduos suscetíveis a agentes halogenados e/ou succinilcolina. A crise aguda pode se manifestar clinicamente com hipertermia, hipercapnia, taquipneia, taquicardia, acidose metabólica, hipercalemia, rigidez muscular e rabdomiólise, sendo uma condição extremamente grave. Ocorre em pacientes de todas as etnias e distribuições geográficas, sendo duas vezes mais comum nos homens e menores de 19 anos de idade (50% dos casos). Se não tratada adequadamente, óbito ocorre em 80%–90% dos pacientes. O tratamento consiste em evitar as crises e abordagem seguindo protocolos rígidos nas crises agudas, incluindo-se uso de dantrolene de sódio (reduz a liberação de cálcio do retículo sarcoplasmático do músculo estriado esquelético por limitar a ativação do receptor de rianodina RYR1).2,3
A prevalência da crise de HM é variável, de 1:10.000 em crianças a 1:50.000 em adultos. A frequência relacionada a procedimentos anestésicos varia de 1 em 10.000 a 1 em 250.000, dependendo da população e dos critérios diagnósticos. A suscetibilidade à HM tem sido associada a genes ligados ao metabolismo do cálcio e que codificam proteínas do complexo de acoplamento excitação-contração do músculo esquelético, com uma frequência de 1:217–1:2750 na população geral. O principal gene implicado na suscetibilidade à HM é o RYR1, responsável por aproximadamente 75% dos casos geneticamente confirmados. Famílias raras apresentam variantes nos genes CACNA1S (1%), STAC3 (<1%) ou ASPH.
A apresentação clínica entre portadores de variantes do RYR1 é altamente variável, e o panorama genético é marcado por alta heterogeneidade alélica, penetrância incompleta e expressividade variável. Variantes patogênicas nesse gene resultam em liberação desregulada de cálcio do retículo sarcoplasmático, levando à contração muscular sustentada e crise metabólica.
Devido à heterogeneidade genética e à possibilidade de herança poligênica, o padrão ouro no diagnóstico de suscetibilidade à HM é o teste de contratilidade in vitro fenotípica (IVCT) para o grupo europeu ou o teste de contratilidade com cafeína-halotano (CHCT) para o grupo norte-americano. Atualmente, apenas um teste de contratilidade negativo pode excluir a suscetibilidade à HM, enquanto um teste genético negativo não o faz. No entanto, esforços têm sido feitos para melhorar a capacidade de detecção do teste molecular, que poderia ser uma ferramenta diagnóstica menos invasiva. Por outro lado, variantes no gene RYR1 são muito frequentes na população, e a identificação e classificação dessas variantes como patogênicas requerem testes adicionais e curadoria. Apenas 72 variantes patogênicas ou provavelmente patogênicas no RYR1 são reconhecidas de acordo com a lista do Grupo Europeu de Hipertermia Maligna (EMHG) ( https://www.emhg.org/genetic-scoring-matrix ; acessado em 18 de agosto de 2025), e, além dos critérios do EMHG, existem os critérios do painel de especialistas em curadoria de variantes do ClinGen MHS (VCEP). Variantes missense são as mais comuns, enquanto inserções e duplicações representam menos de 10%. Os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína.
Cada país ou região apresenta diferenças no tipo e na frequência de variantes associadas à HM. A frequência de variantes relatadas em pacientes com HM varia de 37% a 87,5%, sendo as variantes RYR1 p.R614 e p.G2434R as mais frequentes. No entanto, não existem dados genéticos populacionais da América do Sul, exceto pela descrição de casos isolados e famílias. Em estudo recente, desenvolvido na Unifesp – Escola Paulista de Medicina, foram revisados os dados clínicos e laboratoriais de todas as famílias encaminhadas para avaliação na Unidade Brasileira de HM devido a histórico pessoal ou familiar de HM durante anestesia. Foram coletados dados demográficos e clínicos, bem como níveis séricos de creatina quinase (CK), resultados do teste de contratilidade in vitro (TCIV) e resultados de estudos anatomopatológicos do músculo esquelético. A análise molecular foi realizada por meio de sequenciamento de exoma completo (NGS). Pacientes com e sem variantes foram comparados. Variantes no gene RYR1 foram encontradas em 38 pacientes (62,2%), e nenhuma variante foi identificada em 20 pacientes (32,7%). Mais de uma variante no RYR1 foi encontrada em seis indivíduos. Variantes no gene CACNA1S foram encontradas em três pacientes (4,9%), todos com variantes concomitantes no RYR1. Três pacientes apresentaram variantes no gene STAC3 (4,9%). Comparando os grupos de pacientes com variantes no RYR1 com o grupo sem variantes nesse gene, observou-se que o primeiro grupo apresentou valores séricos mais elevados de CK, maior frequência de ptose, estrabismo, e maior amplitude de contratura no TCIV após a administração de cafeína ou halotano. Nesta avaliação preliminar de indivíduos brasileiros com histórico de hipertermia maligna, a frequência de variantes no RYR1 foi semelhante à de relatos anteriores em outros países, porém houve maior frequência de variantes nos genes STAC3 e CACNA1S.4
Também em estudo da Unifesp, foi identificada uma variante rara: duplicação no gene RYR1 na variabilidade do fenótipo de susceptibilidade à Hipertermia Maligna, em uma família com dois irmãos afetados portadores de uma inserção de 18 pares de bases no éxon 91 do gene RYR1, resultando em uma duplicação em fase de 6 aminoácidos (c.12835_12852 dupGAGGGCGCGGCGGGGCTC: 162 p.G4279_T4284insAAGLEG). A expressão relativa do mRNA do gene RYR1 no músculo dos dois pacientes identificou uma redução de aproximadamente 50%, sugerindo um possível alelo hipomórfico. Este achado levanta a questão que os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína. Este estudo reforça a possibilidade de que a duplicação nessa região possa causar defeitos estruturais e um fenótipo mais grave nos pacientes.5
A Rabdomiólise é uma condição potencialmente fatal que envolve a rápida dissolução do músculo esquelético em resposta a uma variedade de fatores desencadeantes, clinicamente caracterizada por um aumento súbito e acentuado, seguido de uma queda nos valores séricos de creatina quinase (CK) As causas mais comuns de rabdomiólise são lesões por esmagamento secundárias a traumas, esforço físico extremo e miopatias metabólicas. As principais características incluem dor muscular e uma elevação súbita e transitória dos valores séricos de CK. A rabdomiólise grave é frequentemente acompanhada por aumento da excreção urinária de mioglobina (mioglobinúria), o que pode levar à insuficiência renal aguda (IRA) e a uma crise metabólica potencialmente fatal. A ampla gama de complicações (por exemplo, insuficiência renal aguda, arritmias cardíacas, síndrome compartimental, coagulação intravascular disseminada) enfatiza a relevância clínica da rabdomiólise em diversas especialidades médicas.
Estudos de coorte retrospectivos focados em rabdomiólise em pacientes hospitalizados na era pré-sequenciamento de nova geração (NGS) concentraram-se particularmente em fatores desencadeantes externos como a principal causa de um evento de rabdomiólise. Esses estudos identificaram toxinas exógenas (drogas ilícitas, álcool), trauma muscular direto, infecções e exercícios extenuantes (Rabdomiólise por Esforço) como alguns dos fatores desencadeantes mais comuns. Exames genéticos de última geração têm associado mais de 30 genes a uma maior suscetibilidade à rabdomiólise. Contudo, um desafio fundamental na abordagem diagnóstica reside na consideração de quais pacientes necessitam de triagem genética diagnóstica após um episódio de rabdomiólise para identificar uma doença neuromuscular ou metabólica pauci-sintomática ou assintomática. Dentre os genes relacionados, destaca-se o RYR1 e variantes nele podem ser responsáveis por uma proporção substancial de pacientes que apresentam sintomas inexplicáveis de rabdomiólise e/ou mialgia por esforço.6
Com o intuito de revisar a abordagem diagnóstica genética da rabdomiólise, uma pesquisa na Holanda foi realizada, tendo como palavra chave o acrônimo 'RHABDO': Recurrent episodes (episódios recorrentes); HyperCKaemia persisting 8 weeks after the event (aumento de CK persistente após 8 semanas do evento); Accustomed exercise—the intensity of the exercise cannot sufficiently explain the rhabdomyolysis event (exercício de costume – a intensidade do exercício não explica por si o evento de rabdomiólise); Blood CK > 50× the upper limit of normal (ULN) or >10,000 IU/L in female Caucasian patients (aumento de CK > 50x ou > 10 000 UI/L em mulheres); Drugs/medication and other exogenous triggers are insufficient to explain the event (drogas/medicações e outros agentes exógenos são insuficientes para explicarem o evento); and Other affected family members or other exertional symptoms (e.g., severe muscle cramps or swelling) (outros familiares afetados ou com sintomas relacionados ao exercício físico). O acrônimo foi baseado em uma revisão da literatura e em nossa experiência clínica e, portanto, atingiu o nível de evidência de opinião de especialistas. A relevância do RHABDO é ainda mais reforçada por um recente workshop do Centro Neuromuscular Europeu (ENMC) envolvendo 21 médicos e pesquisadores de 12 países diferentes, que enfatizou a necessidade de pesquisa coordenada nesta área. Neste estudo retrospectivo bicêntrico, 122 pacientes foram incluídos. Os fatores desencadeantes mais frequentemente relatados que contribuíram para eventos de rabdomiólise foram exercício (72%), febre/infecção (22%) e/ou medicação (18%). Eles foram submetidos a avaliação genética através de painéis genéticos relacionados à miopatia metabólica (82%), sequenciamento de Sanger (49%) e sequenciamento de exoma completo (NGS) (24%), dos quais 52 pacientes (43%) foram submetidos a múltiplos métodos. Uma variante (provavelmente) patogênica foi identificada em 13 pacientes (11%), todos com ≥2 características de RHABDO presentes. O valor preditivo positivo para ≥2 características foi de 14%, enquanto o valor preditivo negativo foi de 100%. Variantes no gene RYR1 foram descritas em quatro pacientes, um relacionado com esforço (RE).7
A Rabdomiólise por Esforço (RE) é uma degradação muscular patológica associada à atividade física extenuante e agravada por múltiplos fatores de risco. Estes incluem baixo nível de condicionamento físico, alto índice de massa corporal, infecção viral em curso e altitude e temperatura elevadas. A sua incidência é de aproximadamente 36,5 por 100.000 pacientes-ano em atletas ou uma taxa semelhante em militares. De modo geral, as diferenças fundamentais entre a RE e outras formas de rabdomiólise residem em suas causas, fatores desencadeantes e populações afetadas. A patologia celular da lesão por esforço repetitivo (LER) centra-se na ruptura da integridade das células musculares, particularmente no que diz respeito à homeostase iônica e à produção de energia. Exercícios extenuantes ou incomuns podem causar lesão direta ao sarcolema e/ou levar à falha na produção de energia, comprometendo a função de bombas iônicas essenciais, como a Na + /K + -ATPase e a Ca2 + -ATPase. Esse comprometimento aumenta a permeabilidade celular aos íons sódio, resultando em um influxo significativo de cálcio (Ca2 +) para as fibras musculares. O aumento da concentração intracelular de cálcio ativa enzimas dependentes de cálcio, incluindo proteases e fosfolipases, que iniciam a destruição de proteínas miofibrilares, citoesqueléticas e de membrana. Esse processo leva à necrose das fibras musculares, liberando conteúdos intracelulares como CK, mioglobina e eletrólitos no fluido extracelular e na circulação sanguínea. O ciclo vicioso resultante envolve contração muscular sustentada devido ao aumento do cálcio, o que esgota ainda mais as reservas de energia e exacerba o dano muscular.
Embora a via celular geral envolvendo sobrecarga de cálcio e depleção de energia, seja compreendida como o mecanismo de dano às células musculares, os mecanismos subjacentes específicos da rabdomiólise (RB) não são universalmente compreendidos. Estudos em modelos animais, como cavalos suscetíveis à RB recorrente, utilizaram com sucesso a análise do transcriptoma (RNA-seq ou análise de microarray) para revelar alterações na expressão gênica em vias relacionadas à regulação do cálcio, estresse oxidativo e função mitocondrial, demonstrando que essas alterações moleculares podem persistir mesmo entre os episódios. No entanto, as fontes fornecidas não oferecem uma visão abrangente de pesquisas semelhantes em larga escala sobre o transcriptoma, conduzidas especificamente em populações humanas com RB. Um estudo elegante com sequenciamento de RNA em amostras de músculo esquelético de 19 pacientes humanos com histórico de RE, coletadas no mínimo seis meses após o evento de RE mais recente, e oito controles saudáveis para investigar o perfil transcriptômico da RE revelou uma forte supressão da função mitocondrial. Essa supressão incluiu as vias da “cadeia de transporte aeróbico de elétrons” e da “fosforilação oxidativa”, indicando comprometimento da produção de energia. Por outro lado, houve uma regulação positiva de genes associados à adesão e às vias relacionadas à matriz extracelular (aumento do desenvolvimento da matriz extracelular), indicando restauração ativa da função muscular em casos de RE meses após o evento agudo.8
Em síntese, destacamos o que estas pesquisas nos ensinam:
Como o gene RYR1 afeta a função muscular
Associação de variantes (mutações) no RYR1 e Hipertermia Maligna (HM)
Como o defeito no RYR1 leva à Rabdomiólise
O que acontece durante a Rabdomiólise por Esforço (RE)
O que os indivíduos com variantes no RYR1 precisam saber
Hipótese
Referências Bibliográficas:
Buscando estar sempre conectado com as notícias relacionadas e de interesse dos portadores de mutação no gene RYR1, mais especificamente a Miopatia Congênita Centronuclear, que se trata da doença que me acomete, tomei conhecimento da recente pré-publicação científica, intitulada, “O Propofol liga-se diretamente e inibe o receptor 1 de rianodina do músculo esquelético (RYR1)”, versão postada em 12 de janeiro de 2024, na qual os pesquisadores, Thomas T. Joseph, MD, PhD¹, Weiming Bu, PhD¹, Omid Haji-Ghassemi, PhD², Yu Seby Chen, PhD², Kellie Woll, PhD², Paul D. Allen, MD, PhD⁴, Grace Brannigan, PhD³, Filip Van Petegem, PhD², Roderic G. Eckenhoff, MD¹, através de resultados obtidos em estudos e ensaios sugerem em seus relatos que o propofol, um agente anestésico intravenoso de curta ação, em concentrações clínicas, se liga ao receptor de rianodina tipo 1 (RYR1), inibindo sua abertura, podendo assim, prevenir as manifestações clínicas da Hipertermia Maligna (HM), mesmo com exposição a agentes desencadeantes como os anestésicos voláteis.
Confira a integra da publicação pelo seguinte link: https://www.biorxiv.org/content/10.1101/2024.01.10.575040v1.full
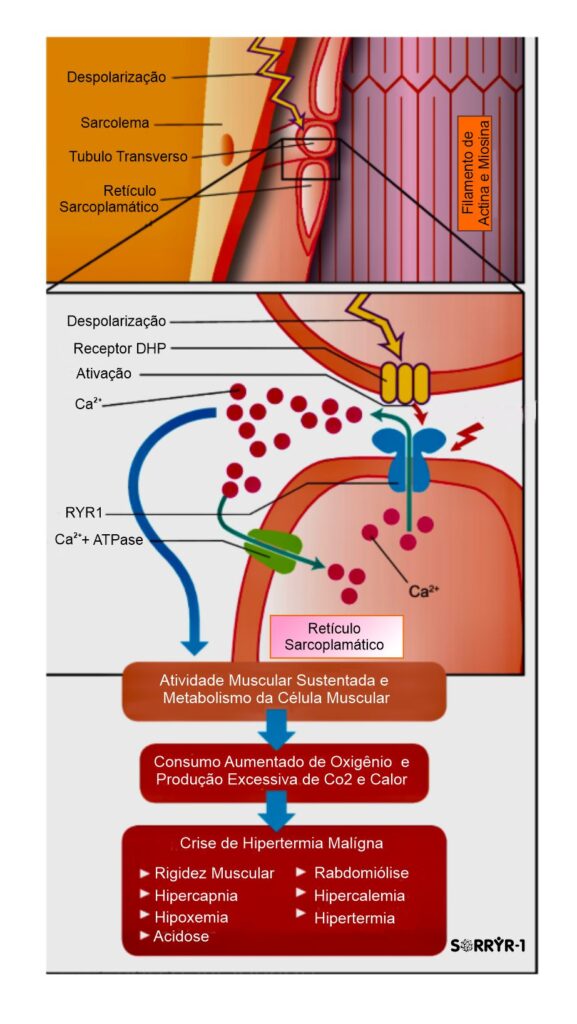
O receptor de rianodina tipo 1 (RYR1) desempenha um papel central na determinação de quando (tempo), e quanta (quantidade) força é produzida pelos músculos esqueléticos, que é necessária e essencial para movimentação do corpo e atividades físicas diárias dos indivíduos. Como principal canal de liberação de íon de cálcio (Ca²⁺) no retículo sarcoplasmático do músculo esquelético, a mutação genética no receptor de rianodina tipo 1 (RYR1), tem a ela subjacentes, algumas doenças ou distúrbios musculares, tais como a Miopatia Centronuclear, Miopatia Central Core, Miopatia Multi-Minicore, Desproporção Congênita do Tipo de Fibra, incluindo distúrbios como a Rabdomiólise por Esforço, e a Hipertermia Maligna (HM).
Em pacientes com a mutação no gene RYR1, “a crise” de Hipertermia Maligna, causada pela exposição a algumas drogas desencadeantes, como os anestésicos voláteis halogenados, pode direcionar o RYR1 a deixar o canal de rianodina em um estado aberto, resultando em uma liberação descontrolada de Ca²⁺, acarretando em tensão no sarcômero, e consequente produção de calor. A restauração de Ca²⁺ no retículo sarcoplasmático também consome ATP (adenosina trifosfato), molécula responsável pelo depósito de energia celular, gerando também por consequência uma carga metabólica adicional insustentável.
Ao anestesiar pacientes com mutações genéticas conhecidas pela suscetibilidade a Hipertermia Maligna, o anestésico geral intravenoso não desencadeante propofol é comumente substituído por anestésicos desencadeantes. As evidências de ligação direta de agentes anestésicos ao RYR1 ou seus parceiros de ligação são escassas, e as interações em nível atômico do propofol com o RYR1 são totalmente desconhecidas. Os pesquisadores mostram no trabalho acima descrito que o propofol diminui a abertura do receptor do canal de rianodina (RYR1) com vesículas no retículo sarcoplasmático e bicamadas lipídicas planas, e que inibe a liberação de Ca²⁺ induzida por ativador do retículo sarcoplasmático no músculo esquelético humano. Além de confirmar a ligação direta, a marcação por fotoafinidade usando m-azipropofol (AziPm) revelou vários supostos locais de ligação de propofol no RYR1. A projeção pela simulação dinâmica da afinidade de ligação molecular sugere que o propofol se liga a pelo menos um destes locais em concentrações clínicas. Esses achados convidam à hipótese de que, além de o propofol não desencadear a Hipertermia Maligna, ele também pode ser protetor contra a Hipertermia Maligna, inibindo o fluxo induzido de Ca²⁺ através do canal de rianodina - RYR1.
¹ Department of Anesthesiology and Critical Care, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA USA; ² Department of Biochemistry, University of British Columbia, Vancouver, BC, Canada; ³ Department of Physics and Center for Computational and Integrative Biology, Rutgers University, Camden, NJ USA; ⁴ Department of Anesthesiology, University of Tennesee, Knoxville, TN USA
O artigo acima mencionado foi publicado na bioRxiv, repositório aberto de pré-publicação direcionado as ciências biológicas (https://www.biorxiv.org/), e hospedado pelo Cold Spring Harbor Laboratory (CSHL).
Recentemente me submeti a uma colonoscopia e endoscopia no Hospital Israelita Albert Einstein, um exame de rotina, mas dado a minha questão com a suscetibilidade a Hipertermia Maligna, os procedimentos foram feitos cercados por forte critérios de segurança, contudo, infelizmente dado ao pouco conhecimento sobre essa doença, esse protocolo de seguranaça é observado em poucas instituições hospitalar.
A Hipertermia Maligna (HM) é uma condição rara e séria, a qual pode ser desencadeada pela administração de certos anestésicos. Os indivíduos com mutação no gene RYR1 têm a  suscetibilidade em ter o episódio de HM. O episódio de HM se não atendido no momento certo, com as drogas e procedimentos corretos pode ser fatal. É muito importante destacar que atualmente já existem identificados anestésicos que devem ser evitados, contudo, por ser uma doença pouco conhecida, além de se evitar essas drogas, os cuidados em qualquer procedimento médico envolvendo anestesia, deve ter uma atenção dobrada. A aplicação de anestesia não se restringe a procedimentos cirúrgicos, mas também em entubação, pequenas intervenções em pronto-socorro, dentre outros.
suscetibilidade em ter o episódio de HM. O episódio de HM se não atendido no momento certo, com as drogas e procedimentos corretos pode ser fatal. É muito importante destacar que atualmente já existem identificados anestésicos que devem ser evitados, contudo, por ser uma doença pouco conhecida, além de se evitar essas drogas, os cuidados em qualquer procedimento médico envolvendo anestesia, deve ter uma atenção dobrada. A aplicação de anestesia não se restringe a procedimentos cirúrgicos, mas também em entubação, pequenas intervenções em pronto-socorro, dentre outros.
Diante do exposto, se alguém tem suscetibilidade à Hipertermia Maligna, é crucial comunicar isso à equipe médica antes da anestesia. Seguem alguns cuidados a serem considerados:
Dado à seriedade deste assunto, devo iniciar o texto com as palavras conclusivas sobre o tema… a HIPERTERMIA MALIGNA é uma doença grave, com risco de morte, que se apresenta em forma de crise, acionada por um gatilho, que é normalmente uma droga anestésica, sendo os portadores de mutação no gene RYR1, os indivíduos com maior risco de ser afetado, daí a afirmação, “HIPERTERMIA MALIGNA, PONTO DE ATENÇÃO PARA OS MÉDICOS ANESTESISTAS, PREOCUPAÇÃO PARA OS PORTADORES DE MUTAÇÃO DO GENE RYR1”. Os anestesistas devem ter o conhecimento sobre a doença e assumir que todas as pessoas com mutação no gene RYR1 correm risco de Hipertermia Maligna. No caso de qualquer procedimento médico que seja necessário anestesia, o portador de mutação de no gene RYR1 deve fazer com que o cirurgião e anestesista saiba sobre sua mutação no gene RYR1, portanto com Suscetibilidade a Hipertermia Maligna (MHS), assumindo assim os eventuais riscos, para tomar precauções e administrar um tipo seguro de anestésico. É recomendado também aos portadores da mutação do RYR1 que portem uma identificação de advertência médica indicando seu risco de HM, em caso de emergência, neste caso, um adesivo em todos os documentos pessoais (Ex.: Identidade, CNH, Passaporte, Carteira do Plano de Saúde, e etc).
CONCEITO
A Hipertermia Maligna (HM) é uma patologia de relação farmacogenética. E isso significa que a doença se manifesta através de um episódio/crise em indivíduos que tenham uma suscetibilidade genética, devido a uma mutação em um determinado gene, acontecendo caso sejam expostos a gatilhos anestésicos (fármacos = medicamentos). Essa predisposição é chamada de "Suscetibilidade à Hipertermia Maligna” (MHS). Genericamente, explica-se que a Hipertermia Maligna (HM) é uma reação biológica em que o corpo humano superaquece a ponto de um colapso muscular, e é considerada uma emergência médica. Se alguém com Hipertermia Maligna não for tratado a tempo, como consequência pode resultar em insuficiência renal, dano cerebral, parada cardíaca, falência de órgãos adicionais, e até morte.
Os indivíduos com Suscetibilidade à Hipertermia Maligna (MHS) podem também apresentar uma crise em resposta a outros gatilhos externos, como por exemplo, ao esforço físico, que poderá causar a Rabdomiólise (quebra muscular), e neste caso apresentando outros sintomas, tais como, cãibras severas, rigidez muscular, e intolerância ao calor.
A genética à Suscetibilidade à Hipertermia Maligna (MHS) é complexa, e vários genes têm sido identificados desempenhando um papel patogênico na Hipertermia Maligna, sendo o RYR1 o mais estudado. Este gene codifica o receptor 1 de Ryanodina (RyR-1), uma proteína do canal de cálcio do retículo sarcoplasmático, expressa predominante na célula muscular. Sabe-se que na maioria dos casos da Suscetibilidade à Hipertermia Maligna (MSH), ela apresenta um traço autossômico dominante, e isso significa que se você tem MHS, um de seus pais provavelmente também tem a MHS. Também significa que cada um de seus filhos têm 50% de chance de herdar a MHS. No entanto, a dita complexidade se prova quando os médicos também observaram a Hipertermia Maligna em pessoas com mutações RYR1 autossômicas recessivas.
Em alguns casos, ao contrário do que se pensa, a Suscetibilidade à Hipertermia Maligna (MHS) pode ocorrer na ausência de fraqueza muscular, em outras palavras, os indivíduos com MHS têm força normal ou até mesmo aumentada, e seu único “sintoma” é a suscetibilidade a reações de Hipertermia Maligna (HM). Por outro lado, a MHS também pode ocorrer em pacientes com Doenças Relacionadas ao RYR1 (RYR-1-RD) com sinais e sintomas típicos de miopatia (fraqueza muscular). Os gatilhos para Hipertermia Maligna incluem certos medicamentos usados para anestesia geral, ou seja, quando alguém é “colocado para dormir”, geralmente antes de uma cirurgia. A anestesia geral é usada em uma ampla variedade de ambientes, incluindo salas de cirurgia, salas de emergência e unidades de terapia intensiva (UTI). Medicamentos específicos conhecidos por desencadear a Hipertermia Maligna incluem anestésicos administrados por via intravenosa, e inalatória via tubo respiratório, como segue:
Mutações no RYR1 associadas a Hipertermia Maligna (HM) mostram uma penetrância variável, e isso significa que uma pessoa pode passar por várias exposições a gatilhos sem problemas antes que uma reação de HM ocorra pela primeira vez. Para tornar as coisas ainda mais confusas, as pessoas com a mesma mutação (incluindo membros da mesma família) podem ter reação clínica diferente, o que significa que algumas podem ser sensíveis ao calor, algumas podem ter reações de HM à anestesia, algumas podem ter rabdomiólise com exercícios físicos, e algumas podem não ter problemas com nenhuma dessas condições.
QUADRO CLÍNICO
O quadro clínico de um episódio/crise de Hipertermia Maligna é variável, e compreende manifestações de alterações metabólicas, de lesão muscular, e das complicações secundárias. Esta condição é expressa por rigidez muscular, aumento do consumo de oxigênio e produção de gás carbônico, acidemia (respiratória e metabólica), taquicardia, taquipnéia, hiperpotassemia, rabdomiólise e mioglobinúria. A dessaturação da hemoglobina no sangue arterial pode ser identificada por oximetria de pulso. Entre os diversos fatores que potencialmente contribuem para a dessaturação persistente, encontram-se acidemia, hipercarbia e hipertermia, capazes de deslocar a curva de saturação da hemoglobina para a direita. A hipercarbia, já detectada na cartografia, parece preceder as demais manifestações. A forma fulminante da Hipertermia Maligna é caracterizada por hipercapnia, rigidez muscular, hipertermias graves, e rabdomiólise, mas situações como cirurgias cardíacas sob circulação extracorpórea (CEC) com hipotermia podem atenuar a expressão clínica da Hipertermia Maligna (HM). A hiperventilação pode mascarar o diagnóstico de HM. Bloqueadores neuromusculares podem retardar o início das manifestações da crise de HM. Convém destacar que nem sempre hipertermia é manifestação inicial ou proeminente da HM. A rigidez muscular pode inexistir em 25 % dos casos, e a Hipertermia ser registrada em apenas um terço deles. A HM surge a qualquer momento durante a anestesia, tendo sido descrita sua ocorrência até 3 horas após a interrupção da exposição ao agente desencadeante (gatilho). A crise de Hipertermia Maligna (HM) pode manifestar-se tardiamente, mesmo após a interrupção da administração do agente desencadeante (gatilho), talvez a imobilidade determinada pela própria anestesia limite a liberação de cálcio a partir do retículo sarcoplasmático. Ao acordar, aumenta a atividade muscular e, na presença de resíduos anestésicos, vêm-se potencializadas à liberação intracelular de cálcio e seus efeitos metabólicos. Tem-se a impressão de haver diferenças entre os halogenados com relação ao seu potencial para desencadear crises de Hipertermia Maligna (HM). O halotano parece ser o de maior risco. A exposição ao isoflurano pode associar-se à crise de HM de início tardio. Parece que a indução da liberação de cálcio do retículo sarcoplasmático pelo sevoflurano é menos intensa em comparação aos demais agentes.
Observação de Manifestações Clínicas Iniciais: Taquicardia - 96,0%, Rigidez muscular - 83,6%, Instabilidade hemodinâmica - 85,5%, Taquipnéia - 85,0%, Cianose - 71,1%, Hipertermia - 30,0%
TERAPIA
Dada a gravidade da doença, o tratamento da Hipertermia Maligna no regime operatório deve ser iniciado em caráter de emergência. A administração do anestésico deve ser interrompida imediatamente, e o paciente deve então receber infusão intravenosa de dantroleno sódico, um relaxante muscular que restaura os níveis fisiológicos de cálcio nos músculos.
Posteriormente, o paciente é induzido a baixar a temperatura corporal por meio de fluidos frios e bolsas de gelo para evitar consequências no cérebro, e é administrado oxigênio para satisfazer o aumento da demanda do organismo. Além disso, a acidose metabólica induzida por lactato é prontamente tratada e o desequilíbrio eletrolítico corrigido. O sucesso da intervenção depende em grande parte da rapidez no reconhecimento dos sintomas e da resposta individual do paciente à terapia.
Nos últimos trinta anos, graças a novos estudos e descobertas no campo farmacológico, a taxa de mortalidade da hipertermia maligna caiu drasticamente, de 70-80 % para 5%, tornando-se uma doença relativamente manejável e tratável.

