O gene RYR1, descrito em 1989 por MacLennan é responsável por dar as instruções para a proteína do receptor de rianodina 1 (RYR1), que é um canal de cálcio crucial para a contração e relaxamento dos músculos esqueléticos.1 A função principal do gene é codificar o canal que libera íons de cálcio do retículo sarcoplasmático (um reservatório dentro da célula muscular) quando o músculo é ativado por um sinal elétrico. Quando o canal RYR1 se abre, o cálcio entra no citoplasma, permitindo que os músculos se contraiam para permitir o movimento. Variantes (mutações) neste gene podem levar a doenças musculares como miopatias congênitas: Doença do Núcleo Central (Central Core), Doença Multi-Minicore, Miopatia Centronuclear e Desproporção Congênita do Tipo de Fibra. O estudo destas doenças permitiu o sequenciamento do gene RYR1 e a descoberta da relação de variantes neste gene e manifestações clínicas como Hipertermia Maligna (HM), Rabdomiólise e Rabdomiólise por Esforço (RE).
RYR1 está localizado no cromossomo 19q13.1 e codifica o Receptor de Rianodina Tipo 1 (RYR1). O gene RYR1 compreende 106 exons e codifica uma grande proteína com 5038 aminoácidos. Devido ao seu tamanho e complexidade consideráveis, o RYR1 tem sido historicamente um gene desafiador para estudo. Durante muitos anos, os esforços de pesquisa concentraram-se principalmente em três domínios (N-terminal, central e C-terminal) considerados pontos críticos de mutação. No entanto, com o advento das tecnologias de Sequenciamento de Nova Geração (NGS), tornou-se possível analisar o gene inteiro. Como resultado, inúmeras novas variantes foram identificadas fora dos pontos críticos previamente conhecidos. Até o momento, mais de 1000 variantes distintas do gene RYR1 foram relatadas (HGMD; https://www.hgmd.cf.ac.uk/ (acessado em 04 de dezembro de 2025) e LOVD; https://databases.lovd.nl/shared/genes/RYR1 (acessado em 04 de dezembro de 2025), porém apenas 72 foram classificadas como variantes diagnósticas pelo Grupo Europeu de Hipertermia Maligna (EMHG; https://www.emhg.org/diagnostic-mutations (acessado em 04 de dezembro de 2025)). A grande maioria dessas variantes são de sentido trocado, enquanto inserções, deleções e duplicações são relativamente incomuns, representando menos de 10% de todas as variantes patogênicas conhecidas no gene RYR1.
A Hipertermia Maligna (HM) é uma síndrome hipermetabólica farmacogenética que se manifesta como uma crise aguda após a exposição de indivíduos suscetíveis a agentes halogenados e/ou succinilcolina. A crise aguda pode se manifestar clinicamente com hipertermia, hipercapnia, taquipneia, taquicardia, acidose metabólica, hipercalemia, rigidez muscular e rabdomiólise, sendo uma condição extremamente grave. Ocorre em pacientes de todas as etnias e distribuições geográficas, sendo duas vezes mais comum nos homens e menores de 19 anos de idade (50% dos casos). Se não tratada adequadamente, óbito ocorre em 80%–90% dos pacientes. O tratamento consiste em evitar as crises e abordagem seguindo protocolos rígidos nas crises agudas, incluindo-se uso de dantrolene de sódio (reduz a liberação de cálcio do retículo sarcoplasmático do músculo estriado esquelético por limitar a ativação do receptor de rianodina RYR1).2,3
A prevalência da crise de HM é variável, de 1:10.000 em crianças a 1:50.000 em adultos. A frequência relacionada a procedimentos anestésicos varia de 1 em 10.000 a 1 em 250.000, dependendo da população e dos critérios diagnósticos. A suscetibilidade à HM tem sido associada a genes ligados ao metabolismo do cálcio e que codificam proteínas do complexo de acoplamento excitação-contração do músculo esquelético, com uma frequência de 1:217–1:2750 na população geral. O principal gene implicado na suscetibilidade à HM é o RYR1, responsável por aproximadamente 75% dos casos geneticamente confirmados. Famílias raras apresentam variantes nos genes CACNA1S (1%), STAC3 (<1%) ou ASPH.
A apresentação clínica entre portadores de variantes do RYR1 é altamente variável, e o panorama genético é marcado por alta heterogeneidade alélica, penetrância incompleta e expressividade variável. Variantes patogênicas nesse gene resultam em liberação desregulada de cálcio do retículo sarcoplasmático, levando à contração muscular sustentada e crise metabólica.
Devido à heterogeneidade genética e à possibilidade de herança poligênica, o padrão ouro no diagnóstico de suscetibilidade à HM é o teste de contratilidade in vitro fenotípica (IVCT) para o grupo europeu ou o teste de contratilidade com cafeína-halotano (CHCT) para o grupo norte-americano. Atualmente, apenas um teste de contratilidade negativo pode excluir a suscetibilidade à HM, enquanto um teste genético negativo não o faz. No entanto, esforços têm sido feitos para melhorar a capacidade de detecção do teste molecular, que poderia ser uma ferramenta diagnóstica menos invasiva. Por outro lado, variantes no gene RYR1 são muito frequentes na população, e a identificação e classificação dessas variantes como patogênicas requerem testes adicionais e curadoria. Apenas 72 variantes patogênicas ou provavelmente patogênicas no RYR1 são reconhecidas de acordo com a lista do Grupo Europeu de Hipertermia Maligna (EMHG) ( https://www.emhg.org/genetic-scoring-matrix ; acessado em 18 de agosto de 2025), e, além dos critérios do EMHG, existem os critérios do painel de especialistas em curadoria de variantes do ClinGen MHS (VCEP). Variantes missense são as mais comuns, enquanto inserções e duplicações representam menos de 10%. Os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína.
Cada país ou região apresenta diferenças no tipo e na frequência de variantes associadas à HM. A frequência de variantes relatadas em pacientes com HM varia de 37% a 87,5%, sendo as variantes RYR1 p.R614 e p.G2434R as mais frequentes. No entanto, não existem dados genéticos populacionais da América do Sul, exceto pela descrição de casos isolados e famílias. Em estudo recente, desenvolvido na Unifesp – Escola Paulista de Medicina, foram revisados os dados clínicos e laboratoriais de todas as famílias encaminhadas para avaliação na Unidade Brasileira de HM devido a histórico pessoal ou familiar de HM durante anestesia. Foram coletados dados demográficos e clínicos, bem como níveis séricos de creatina quinase (CK), resultados do teste de contratilidade in vitro (TCIV) e resultados de estudos anatomopatológicos do músculo esquelético. A análise molecular foi realizada por meio de sequenciamento de exoma completo (NGS). Pacientes com e sem variantes foram comparados. Variantes no gene RYR1 foram encontradas em 38 pacientes (62,2%), e nenhuma variante foi identificada em 20 pacientes (32,7%). Mais de uma variante no RYR1 foi encontrada em seis indivíduos. Variantes no gene CACNA1S foram encontradas em três pacientes (4,9%), todos com variantes concomitantes no RYR1. Três pacientes apresentaram variantes no gene STAC3 (4,9%). Comparando os grupos de pacientes com variantes no RYR1 com o grupo sem variantes nesse gene, observou-se que o primeiro grupo apresentou valores séricos mais elevados de CK, maior frequência de ptose, estrabismo, e maior amplitude de contratura no TCIV após a administração de cafeína ou halotano. Nesta avaliação preliminar de indivíduos brasileiros com histórico de hipertermia maligna, a frequência de variantes no RYR1 foi semelhante à de relatos anteriores em outros países, porém houve maior frequência de variantes nos genes STAC3 e CACNA1S.4
Também em estudo da Unifesp, foi identificada uma variante rara: duplicação no gene RYR1 na variabilidade do fenótipo de susceptibilidade à Hipertermia Maligna, em uma família com dois irmãos afetados portadores de uma inserção de 18 pares de bases no éxon 91 do gene RYR1, resultando em uma duplicação em fase de 6 aminoácidos (c.12835_12852 dupGAGGGCGCGGCGGGGCTC: 162 p.G4279_T4284insAAGLEG). A expressão relativa do mRNA do gene RYR1 no músculo dos dois pacientes identificou uma redução de aproximadamente 50%, sugerindo um possível alelo hipomórfico. Este achado levanta a questão que os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína. Este estudo reforça a possibilidade de que a duplicação nessa região possa causar defeitos estruturais e um fenótipo mais grave nos pacientes.5
A Rabdomiólise é uma condição potencialmente fatal que envolve a rápida dissolução do músculo esquelético em resposta a uma variedade de fatores desencadeantes, clinicamente caracterizada por um aumento súbito e acentuado, seguido de uma queda nos valores séricos de creatina quinase (CK) As causas mais comuns de rabdomiólise são lesões por esmagamento secundárias a traumas, esforço físico extremo e miopatias metabólicas. As principais características incluem dor muscular e uma elevação súbita e transitória dos valores séricos de CK. A rabdomiólise grave é frequentemente acompanhada por aumento da excreção urinária de mioglobina (mioglobinúria), o que pode levar à insuficiência renal aguda (IRA) e a uma crise metabólica potencialmente fatal. A ampla gama de complicações (por exemplo, insuficiência renal aguda, arritmias cardíacas, síndrome compartimental, coagulação intravascular disseminada) enfatiza a relevância clínica da rabdomiólise em diversas especialidades médicas.
Estudos de coorte retrospectivos focados em rabdomiólise em pacientes hospitalizados na era pré-sequenciamento de nova geração (NGS) concentraram-se particularmente em fatores desencadeantes externos como a principal causa de um evento de rabdomiólise. Esses estudos identificaram toxinas exógenas (drogas ilícitas, álcool), trauma muscular direto, infecções e exercícios extenuantes (Rabdomiólise por Esforço) como alguns dos fatores desencadeantes mais comuns. Exames genéticos de última geração têm associado mais de 30 genes a uma maior suscetibilidade à rabdomiólise. Contudo, um desafio fundamental na abordagem diagnóstica reside na consideração de quais pacientes necessitam de triagem genética diagnóstica após um episódio de rabdomiólise para identificar uma doença neuromuscular ou metabólica pauci-sintomática ou assintomática. Dentre os genes relacionados, destaca-se o RYR1 e variantes nele podem ser responsáveis por uma proporção substancial de pacientes que apresentam sintomas inexplicáveis de rabdomiólise e/ou mialgia por esforço.6
Com o intuito de revisar a abordagem diagnóstica genética da rabdomiólise, uma pesquisa na Holanda foi realizada, tendo como palavra chave o acrônimo 'RHABDO': Recurrent episodes (episódios recorrentes); HyperCKaemia persisting 8 weeks after the event (aumento de CK persistente após 8 semanas do evento); Accustomed exercise—the intensity of the exercise cannot sufficiently explain the rhabdomyolysis event (exercício de costume – a intensidade do exercício não explica por si o evento de rabdomiólise); Blood CK > 50× the upper limit of normal (ULN) or >10,000 IU/L in female Caucasian patients (aumento de CK > 50x ou > 10 000 UI/L em mulheres); Drugs/medication and other exogenous triggers are insufficient to explain the event (drogas/medicações e outros agentes exógenos são insuficientes para explicarem o evento); and Other affected family members or other exertional symptoms (e.g., severe muscle cramps or swelling) (outros familiares afetados ou com sintomas relacionados ao exercício físico). O acrônimo foi baseado em uma revisão da literatura e em nossa experiência clínica e, portanto, atingiu o nível de evidência de opinião de especialistas. A relevância do RHABDO é ainda mais reforçada por um recente workshop do Centro Neuromuscular Europeu (ENMC) envolvendo 21 médicos e pesquisadores de 12 países diferentes, que enfatizou a necessidade de pesquisa coordenada nesta área. Neste estudo retrospectivo bicêntrico, 122 pacientes foram incluídos. Os fatores desencadeantes mais frequentemente relatados que contribuíram para eventos de rabdomiólise foram exercício (72%), febre/infecção (22%) e/ou medicação (18%). Eles foram submetidos a avaliação genética através de painéis genéticos relacionados à miopatia metabólica (82%), sequenciamento de Sanger (49%) e sequenciamento de exoma completo (NGS) (24%), dos quais 52 pacientes (43%) foram submetidos a múltiplos métodos. Uma variante (provavelmente) patogênica foi identificada em 13 pacientes (11%), todos com ≥2 características de RHABDO presentes. O valor preditivo positivo para ≥2 características foi de 14%, enquanto o valor preditivo negativo foi de 100%. Variantes no gene RYR1 foram descritas em quatro pacientes, um relacionado com esforço (RE).7
A Rabdomiólise por Esforço (RE) é uma degradação muscular patológica associada à atividade física extenuante e agravada por múltiplos fatores de risco. Estes incluem baixo nível de condicionamento físico, alto índice de massa corporal, infecção viral em curso e altitude e temperatura elevadas. A sua incidência é de aproximadamente 36,5 por 100.000 pacientes-ano em atletas ou uma taxa semelhante em militares. De modo geral, as diferenças fundamentais entre a RE e outras formas de rabdomiólise residem em suas causas, fatores desencadeantes e populações afetadas. A patologia celular da lesão por esforço repetitivo (LER) centra-se na ruptura da integridade das células musculares, particularmente no que diz respeito à homeostase iônica e à produção de energia. Exercícios extenuantes ou incomuns podem causar lesão direta ao sarcolema e/ou levar à falha na produção de energia, comprometendo a função de bombas iônicas essenciais, como a Na + /K + -ATPase e a Ca2 + -ATPase. Esse comprometimento aumenta a permeabilidade celular aos íons sódio, resultando em um influxo significativo de cálcio (Ca2 +) para as fibras musculares. O aumento da concentração intracelular de cálcio ativa enzimas dependentes de cálcio, incluindo proteases e fosfolipases, que iniciam a destruição de proteínas miofibrilares, citoesqueléticas e de membrana. Esse processo leva à necrose das fibras musculares, liberando conteúdos intracelulares como CK, mioglobina e eletrólitos no fluido extracelular e na circulação sanguínea. O ciclo vicioso resultante envolve contração muscular sustentada devido ao aumento do cálcio, o que esgota ainda mais as reservas de energia e exacerba o dano muscular.
Embora a via celular geral envolvendo sobrecarga de cálcio e depleção de energia, seja compreendida como o mecanismo de dano às células musculares, os mecanismos subjacentes específicos da rabdomiólise (RB) não são universalmente compreendidos. Estudos em modelos animais, como cavalos suscetíveis à RB recorrente, utilizaram com sucesso a análise do transcriptoma (RNA-seq ou análise de microarray) para revelar alterações na expressão gênica em vias relacionadas à regulação do cálcio, estresse oxidativo e função mitocondrial, demonstrando que essas alterações moleculares podem persistir mesmo entre os episódios. No entanto, as fontes fornecidas não oferecem uma visão abrangente de pesquisas semelhantes em larga escala sobre o transcriptoma, conduzidas especificamente em populações humanas com RB. Um estudo elegante com sequenciamento de RNA em amostras de músculo esquelético de 19 pacientes humanos com histórico de RE, coletadas no mínimo seis meses após o evento de RE mais recente, e oito controles saudáveis para investigar o perfil transcriptômico da RE revelou uma forte supressão da função mitocondrial. Essa supressão incluiu as vias da “cadeia de transporte aeróbico de elétrons” e da “fosforilação oxidativa”, indicando comprometimento da produção de energia. Por outro lado, houve uma regulação positiva de genes associados à adesão e às vias relacionadas à matriz extracelular (aumento do desenvolvimento da matriz extracelular), indicando restauração ativa da função muscular em casos de RE meses após o evento agudo.8
Em síntese, destacamos o que estas pesquisas nos ensinam:
Como o gene RYR1 afeta a função muscular
Associação de variantes (mutações) no RYR1 e Hipertermia Maligna (HM)
Como o defeito no RYR1 leva à Rabdomiólise
O que acontece durante a Rabdomiólise por Esforço (RE)
O que os indivíduos com variantes no RYR1 precisam saber
Hipótese
Referências Bibliográficas:
A Miopatia Congênita Centronuclear é uma doença que causa muitas exclamações, mas também, interrogações. Sempre que as pessoas chegam até mim para questionar sobre minha doença, elas fazem uma exclamação dizendo, “nunca tinha ouvido sobre sua doença !”, e em seguida questionam como ela me afeta, e mais uma vez elas exclamam dizendo, "puxa vida !", e por ultimo exclamam novamente, “essa é uma super rara !”, daí respondo, "sim, no pé da palavra, ela é uma Doença Rara ou talvez uma Doença Ultrarrara.
A Miopatia Congênita Centronuclear decorrente de mutações no gene RYR1 é classificada como Doenças Raras ou Doenças Ultrarraras ?
Entender essas condições é essencial para oferecer o suporte adequado e assegurar abordagens terapêuticas personalizadas tanto para o paciente quanto para seus familiares. Doenças raras nem sempre recebem a atenção que merecem porque afetam relativamente poucas pessoas. Muitas vezes, pode levar anos para que uma pessoa receba o diagnóstico correto de uma doença rara, e cerca de 95% das doenças raras e ultrarraras ainda não têm tratamento.
Diferentes partes do mundo, as doenças raras e ultrarraras são identificadas e tratadas de maneiras diferentes, mas de um modo genérico a definição é mais ou menos assim:
Uma Doença Rara é aquela que acomete no máximo 65 pessoas a cada 100.000 habitantes, o que corresponde a uma prevalência aproximada de 1 para cada 1.500 indivíduos. A maior parte dessas condições tem origem genética, embora algumas possam surgir devido a fatores infecciosos ou relacionados ao sistema imunológico.
A Doença Ultrarrara, como o próprio nome indica, corresponde a condições ainda mais incomuns, com uma incidência de cerca de 1 caso para cada 50.000 indivíduos. Por serem extremamente infrequentes e pouco conhecidas, essas doenças geralmente apresentam maior dificuldade diagnóstica e exigem cuidados altamente especializados e personalizados.
Com relação à questão levantada no início do texto, a prevalência exata da Miopatia Congênita Centronuclear causada especificamente pela mutação no gene RYR-1 é desconhecida.
A dificuldade em determinar um número exato ocorre porque:
• Ela é uma doença ultrarrara por estar dentro de um grupo de doenças já raras, as Miopatias Congênitas.
• Os dados epidemiológicos costumam ser apresentados para a Miopatia Congênita Centronuclear (MCCN) considerando o conjunto das patologias que compõem esse grupo, que inclui mutações nos genes MTM1, DNM2, BIN1 e RYR1.
• O gene RYR1 está associado a um espectro de doenças musculares conhecidas como Doenças Relacionadas ao RYR1 ou RYR1-DR, sendo a forma Centronuclear apenas uma das possíveis manifestações, juntamente com a Miopatia Central Core (MCC), e outras.
Embora não haja um número específico de prevalência para a Miopatia Congênita Centronuclear causada pela mutação no RYR1, podemos contextualizar com base nas informações disponíveis para as condições relacionadas:
• No grupo da Miopatia Congênita Centronuclear (MCCN) em geral, a prevalência geral do grupo é desconhecida. A forma mais comum dentro deste grupo é a Miopatia Miotubular (ligada ao X) causada por mutações no gene MTM1, cuja incidência é estimada em cerca de 1 em 50.000 indivíduos do sexo masculino.
• No grupo das Doenças Relacionadas ao RYR1 (RYR1-DR) em geral, a doença mais frequentemente associado é a Miopatia Central Core (MCC), que é a forma mais comum de miopatia congênita não distrófica.
Um estudo de uma série pediátrica na Espanha estimou a incidência de Miopatias Relacionadas ao RYR1, que abrange a Centronuclear e a Central Core, se mostra em cerca de 1 em 10.000 nascidos vivos na área de estudo.
Em síntese, a mutação no gene RYR1 está associada a um fenótipo de ocorrência rara. A prevalência específica do subtipo Centronuclear (MCCN) ainda não foi estabelecida de forma isolada na literatura médica, sendo considerada extremamente baixa. Entretanto, não há consenso atual sobre classificá-la como no grupo de Doenças Raras ou no grupo de Doenças Ultrarraras.
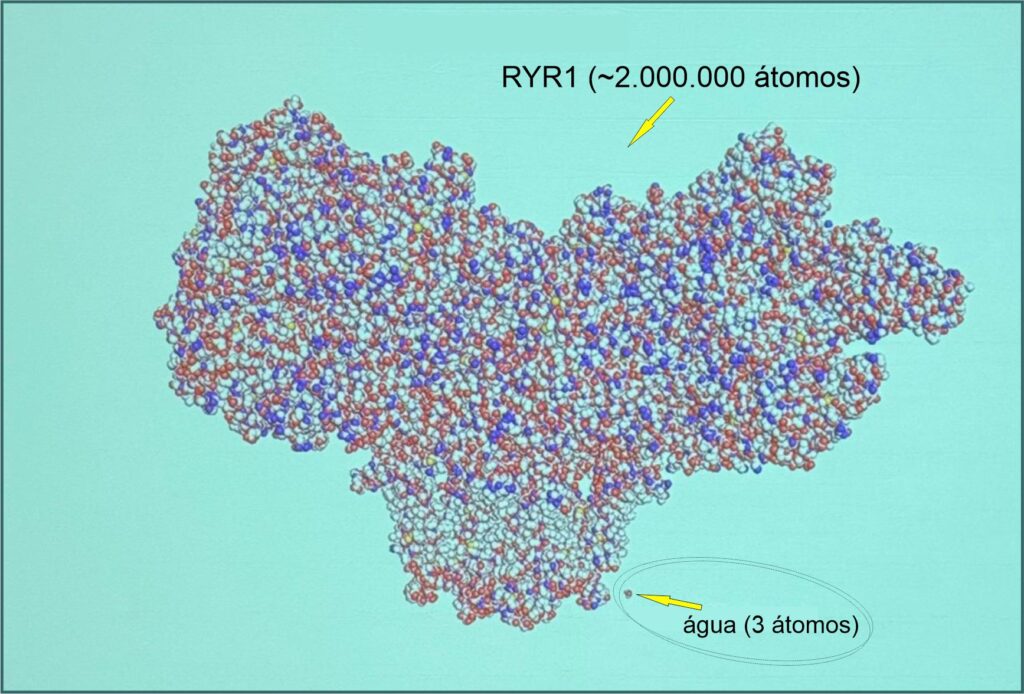
O RYR1 é um dos maiores genes do corpo humano, e é responsável para o funcionamento dos nossos músculos.
O gene RYR1 (Receptor de Rianodina 1) é um dos maiores e mais complexos genes do genoma humano. Ele contém mais de 100 exons e cerca de 15 mil pares de bases na sua sequencia de DNA, abrangendo uma grande extensão no cromossomo 19. A estimativa é que o gene RYR1 tenha cerca de 2 milhões de átomos, e esse é somente um cálculo de aproximação, pois a quantidade exata pode variar dependendo da sequência específica de nucleotídeos.
Para se ter uma idéia da complexidade do RYR1, ele é imensamente maior e mais complexo do que moléculas simples como da água, tão crucial para nossa vida, e a título de comparação, pasmem ! …o RYR1 tem 2 milhões de átomos, e a água, também conhecida como H20, tem somente 3 átomos.
O gene RYR1 que é crucial para a função do receptor de rianodina, que é um canal de cálcio localizado no Retículo 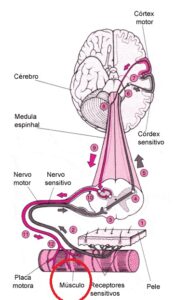 Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+
Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+) do RS para o citoplasma; 4). O cálcio dispara a contração muscular ao permitir a interação Actina-Miosina; 5). Para o relaxamento, o sinal cessa, o RYR1 se fecha, e rapidamente retorna o cálcio do citoplasma, devolvendo-o ao RS.
Em resumo, o RYR1 atua como o principal portão de saída do cálcio do reservatório intracelular. Ele é o mediador essencial que garante que o comando elétrico do nervo seja traduzido em movimento mecânico, controlando a entrada abrupta de cálcio que inicia a contração e, consequentemente, permitindo que a sua reabsorção finalize o processo para o relaxamento.
Como disse no início deste texto, devido ao tamanho e complexidade do RYR1, mutações nesse gene podem estar associadas a várias doenças musculares, incluindo:
O estudo científico do RYR1 é importante para entender essas condições ou doenças, e desenvolver tratamentos adequados em busca de minimizar seus efeitos e sintomas, pensando até na sua própria cura. Além disso, a análise genética pode ajudar em diagnósticos precoces e na prevenção de complicações associadas a essas doenças.
Durante o tempo que tenho acompanhado a movimentação de informações em torno das questões relacionadas ao RYR1, observo um grande esforço da comunidade científica em busca de entender o mecanismo de funcionamento deste complexo e grandioso gene, grande no tamanho, mas sobretudo grande em sua importância. Essas questões fazem com que o desafio ainda seja maior não só por conta dos indivíduos que têm uma doença a ele relacionada, mas também para compreensão sobre o impacto do envelhecimento celular em qualquer outro indivíduo. Observo também um grande movimento de trabalhos sendo desenvolvidos por cientistas de toda parte do mundo em busca de desenvolver uma droga para tratamentos adequados, para de repente ao menos minimizar seus efeitos e sintomas, até sua cura das doenças relacionadas ao RYR1. Particularmente, minha percepção é que as pesquisas através da técnica de reposicionamento de drogas, também chamado de redirecionamento de fármacos têm se mostrado mais promissoras no curto prazo. O “reposicionamento de drogas” se trata de uma estratégia que busca descobrir novas aplicações terapêuticas para medicamentos já existentes, que originalmente foram desenvolvidos para tratar outras doenças. Essa técnica tem a vantagem por seus reduzidos custos, assim como pela redução de tempo gasto no processo de desenvolvimento, e menores riscos no uso da eventual droga, pois já se conhece a farmacocinética e toxicidade do medicamento pesquisado, um exemplo real e prático de pesquisa em curso está se dando com o Sulfato de Salbutamol.
Contudo, a terapia genética também tem se mostrado uma abordagem promissora para o tratamento e cura de diversas doenças genéticas, oferecendo a possibilidade de corrigir ou substituir genes defeituosos. Embora este tipo de tratamento ainda esteja em desenvolvimento e não seja uma solução universal para algumas doenças, como por exemplo certas formas de miopatias ou doenças hereditárias, os resultados das pesquisas têm sido encorajadores. No entanto, a eficácia e a segurança da terapia genética podem variar dependendo da doença, do tipo de terapia utilizada, e condições do paciente. Além disso, questões éticas e de custo também são consideradas importantes. Contudo, enquanto a terapia genética oferece uma expectativa otimista, é uma área em evolução que requer mais pesquisas, altos recursos financeiros, e testes clínicos para se consolidar como a melhor solução para todas as doenças genéticas.
No caso específico das doenças relacionadas à mutação do gene RYR1, a terapia genética apresenta vários desafios, principalmente, como escrito no início deste texto, devido ao tamanho do gene, complexidade das doenças relacionadas a ele, mas também por considerar que os músculos representam aproximadamente 40% da massa total do corpo humano, e a maior parte dessa massa muscular é composta por músculos esqueléticos. Observe a seguir algumas das dificuldades:
Esses desafios tornam a pesquisa e o desenvolvimento de terapias para mutações no gene RYR1 complexos e exigem abordagens inovadoras e multidisciplinares.
Eu, enquanto portador de uma doença causada pela mutação nesse “grandioso” RYR1, tenho uma relação muito particular com esse gene, e a cada dia que passa o conheço um pouco mais, ..... confira minha próxima postagem intitulada “Eu vi o meu gene em 3D e entendi o que acontece dentro de mim”.
A grande maioria das pessoas a partir dos 50 anos se deparam com complicações ortopédicas, seja nas articulações, coluna, quadril, dentre outros, e isso geralmente se deve pelo uso inadequado do corpo gerando os desgastes naturais que acontecem no decorrer da vida, ou acontece também pela falta de exercícios físicos.
No caso do indivíduo portador de miopatia centronuclear, doença essa causada pela mutação no gene RYR-1, responsável pelo funcionamento dos músculos, as implicações ortopédicas em relação a uma pessoa sem a doença, é potencializada, e pode levar a uma série de complicações já desde o nascimento. Como já explicado em postagens anteriores, cada pessoa com doença relacionada ao RYR-1 é única, assim como a evolução da doença, as complicações também podem afetar diferentemente cada indivíduo.
Nesta postagem, vou discorrer sobre alguns dos principais problemas ortopédicos com os quais pessoalmente convivo, e que podem ocorrer com os indivíduos afetados por uma doença relacionada ao RYR-1, que são:
 nas articulações e tendões. No nosso caso, portadores de miopatia, a situação é logicamente agravada, porque como nossos músculos não são utilizados adequadamente, o enfraquecimento muscular é aumentado, resultando em instabilidade nas articulações, e isso pode aumentar o risco de lesões, como distensões ou rupturas de tendões. Além disso, a imobilidade por fraqueza muscular pode levar à rigidez articular e à perda de amplitude de movimento, o que pode causar dor e desconforto. Essa falta de mobilidade muscular também pode contribuir para o desenvolvimento de condições como a artrite, pois as articulações não recebem o movimento necessário para manter a saúde e a lubrificação adequada.
nas articulações e tendões. No nosso caso, portadores de miopatia, a situação é logicamente agravada, porque como nossos músculos não são utilizados adequadamente, o enfraquecimento muscular é aumentado, resultando em instabilidade nas articulações, e isso pode aumentar o risco de lesões, como distensões ou rupturas de tendões. Além disso, a imobilidade por fraqueza muscular pode levar à rigidez articular e à perda de amplitude de movimento, o que pode causar dor e desconforto. Essa falta de mobilidade muscular também pode contribuir para o desenvolvimento de condições como a artrite, pois as articulações não recebem o movimento necessário para manter a saúde e a lubrificação adequada.
Para concluir, eu diria que as complicações ortopédicas acima descritas podem ser inevitáveis aos portadores de miopatias relacionadas ao RYR1, sejam elas por origem congênita, ou em decorrência das adaptações que o indivíduo precisa fazer para conseguir movimentar durante a vida, ou pelas que acontecem com o desgaste natural do envelhecimento. Como portador de Miopatia Congênita Centronuclear, eu convivo com todas as complicações descritas, e de todas as origens de causas. Contudo, vejo que existem formas de se prevenir, postergar o seu surgimento ou agravamento, e até de se mitigar os sintomas das complicações ortopédicas. A educação postural, e o desenvolvimento pessoal de consciência corporal, devem fazer parte do nosso dia a dia. No caso de as complicações já terem se instaladas, o manejo desses problemas geralmente envolve uma combinação de fisioterapia, alongamentos, terapia ocupacional e, em alguns casos as intervenções cirúrgicas para corrigir deformidades ou melhorar a função do corpo. Por fim, é fundamental que os portadores de miopatia centronuclear sejam acompanhados por uma equipe multidisciplinar para abordar essas questões de forma abrangente, preventiva e terapêutica.
As miopatias são doenças que afetam os músculos, e podem se apresentar desde o nascimento até a idade adulta. Quando a miopatia se manifesta no início da vida, é frequentemente referida como miopatia congênita e de origem genética.
O diagnóstico de miopatia em uma criança geralmente envolve uma combinação de exame físico, história clínica, testes laboratoriais, incluindo biópsia e exame genético, além de exames de imagem. O diagnóstico preciso depende da avaliação completa feita por um médico especialista em doenças neuromusculares pediátricas.
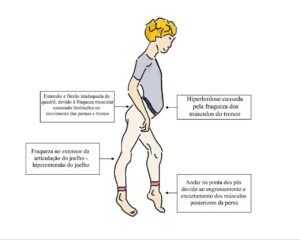
Existem alguns sinais físicos que são característicos em uma criança portadora de miopatia. Além do Movimento de Gowers, já descrito nesta website, o Sinal de Trendelenburg e a Marcha Miopática também são sinais físicos que podem ser cruciais no exame clínico quando do diagnóstico de uma miopatia. Esses sinais são incorporados como característica da doença, podendo ter um impacto significativo na qualidade de vida de quem a sofre, portanto, o acompanhamento e tratamento adequados são essenciais para ajudar os indivíduos a controlar os sintomas, limitações, evitar complicações secundárias, e manter uma boa qualidade de vida.
A Marcha Miopática é um termo médico usado para descrever um padrão de caminhada que é afetado pela fraqueza muscular causada por uma miopatia, que também é conhecida como Marcha do Pato. Essa condição provoca uma série de alterações na maneira de andar dos indivíduos afetados. Um dos sintomas mais característicos é também chamado de marcha bamboleante. Esta marcha é caracterizada pelo balanço do tronco e separação dos pés durante a caminhada. Como os músculos do tronco também são afetados, desencadeia-se uma postura anormal, apresentando como exemplo a hiperlordose lombar, que é uma curvatura excessiva na região lombar. Outra característica distintiva é andar na ponta dos pés, que provoca o espessamento e retração da musculatura posterior da perna, criando uma aparência de pseudo-hipertrofia. Nesta condição, os músculos tornam-se mais espessos e volumosos, mas na verdade é resultado da destruição da fibra muscular e da sua substituição por tecido fibroso ou fibras colágenas. Este padrão de marcha é uma característica comum em várias formas de miopatia e pode variar em gravidade dependendo do tipo e da progressão da doença.
O Sinal de Trendelenburg é uma característica bem comum em indivíduos afetados por uma miopatia, com fraqueza da musculatura abdutora do quadril, em especial o glúteo médio. O nome deste sinal é em homenagem ao cirurgião alemão Friedrich Trendelenburg. O sinal de Trendelenburg é positivo se, quando o quadril de um paciente que está de pé sustentado por somente uma perna, cai para o lado da perna levantada. A fraqueza é presente no lado da perna em contato com o chão. O corpo não é capaz de manter a perna estável, causando uma inclinação ou queda da pelve em comparação com o lado contralateral. Essencialmente, o Sinal de Trendelenburg é causado pela fraqueza dos músculos glúteo médio e mínimo.
O Sinal de Trendelenburg e a Marcha Miopática, apesar de poderem variar dependendo da miopatia, e da forma que ela se apresenta em cada indivíduo, ao longo do tempo, podem desencadear uma série de consequências no corpo (ufa ! …e eu que o diga…), podendo relacionar algumas delas:
Nota: todo o material escrito nesta página é oriundo de pesquisa científica, e conclusões próprias do autor deste website, que convive como portador, desde o nascimento, com a Miopatia Congênita Centronuclear causada pela mutação no gene RYR-1
A terapia genética que era tida como uma futura grande promessa para o tratamento de miopatias relacionadas ao RYR1, se torna uma realidade com a publicação do recente relato científico da primeira correção por Edição Prime de uma mutação no gene RYR1.
A Fundação RYR-1 (https://ryr1.org/) cumprimentou a todos no início de ano com um “Feliz 2024, mas também compartilhou a informação que financiou uma pesquisa incrivelmente importante com o Dr. Jacques P. Tremblay, um pesquisador na Universidade Laval em Quebec. As descobertas e resultados dos trabalhos de pesquisa acabaram de ser publicadas em um novo artigo de  pesquisa (https://www.mdpi.com/2073-4409/13/1/31#). E o resultado é ainda mais emocionante, pois os pesquisadores utilizaram com sucesso a Edição Prime, uma forma de edição genética, que foi utilizada para corrigir uma mutação no gene RYR1 nas células musculares esqueléticas. Esta pesquisa fornece "prova de conceito" para a edição de genes como sendo uma estratégia em potencial para tratar miopatias relacionadas com RYR-1, que atualmente carecem de terapias eficazes. Segundo o cientista, "estes resultados são as primeiras demonstrações de que é possível corrigir mutações no gene RYR-1"
pesquisa (https://www.mdpi.com/2073-4409/13/1/31#). E o resultado é ainda mais emocionante, pois os pesquisadores utilizaram com sucesso a Edição Prime, uma forma de edição genética, que foi utilizada para corrigir uma mutação no gene RYR1 nas células musculares esqueléticas. Esta pesquisa fornece "prova de conceito" para a edição de genes como sendo uma estratégia em potencial para tratar miopatias relacionadas com RYR-1, que atualmente carecem de terapias eficazes. Segundo o cientista, "estes resultados são as primeiras demonstrações de que é possível corrigir mutações no gene RYR-1"
O gene RYR1 codifica um canal de cálcio denominado receptor 1 de Ryanodina, apresentada nas fibras musculares esqueléticas. A falha desse canal causa fraqueza muscular, que degenera acarretando deficiências motoras no indivíduo afetado. Atualmente, não existem tratamentos eficazes para estas miopatias, também conhecidas como doenças relacionadas ao RYR1, que são causadas principalmente por mutações pontuais. A Edição Prime permite a modificação precisa de nucleotídeos no DNA. Os resultados dos trabalhos de pesquisa pelos cientistas Kelly Godbout, Joël Rousseau e Jacques P. Tremblay, demostraram uma taxa de correção de 59% da mutação T4709M no gene RYR1 em mioblastos humanos pela entrega de RNA dos componentes de Edição Prime. Deve-se notar que o T4709M é recessivo e, portanto, as pessoas com mutação heterozigótica são saudáveis. Estes resultados são a primeira demonstração de que é possível corrigir mutações no gene RYR1.
A tecnologia de Edição Prime pode ser usada para corrigir mutações que causam miopatias relacionadas ao RYR1. Este grupo de doenças inclui a Hipertermia Maligna (HM), Miopatia Central Core (CCD), Miopaty Multi-Minicore (MmD), Miopatia Centronuclear (CNM), Desproporção Congênita do Tipo de Fibra (CFTD) e Rabdomiólise por Esforço (ERM). Até o momento, mais de 700 variantes no gene RYR1 foram identificadas. Este gene que codifica uma proteína chamada "receptor de rianodina 1" (RyR1), é o principal canal de cálcio no retículo sarcoplasmático (SR) nas fibras musculares esqueléticas. A disfunção desta proteína afeta o fluxo de cálcio para os músculos. A posição da mutação não afetará ou impactará na proteína, mas as mutações nos genes farão com que ocorra principalmente a um vazamento de cálcio. E como o cálcio é fundamental para a contração muscular, essa desregulação do RYR1 leva à fraqueza muscular, caibras, exaustão, intolerância ao calor, dificuldades respiratórias e até mesmo à reação maligna de hipertermia, ou Hipertermia Maligna. Essas miopatias, portanto, afetam gravemente a qualidade de vida dos pacientes. A proteína RYR1 tem variações funcionais limitadas, e o gene RYR1 é um dos mais intolerantes a variações de sequência no genoma humano.
Até o momento, não existe tratamento eficaz para essas doenças relacionadas ao RYR1. Como muitas mutações nos genes RYR1 são mutações pontuais, os resultados descritos no referido artigo demonstram claramente que a Edição Prime pode ser utilizada para corrigi-las, uma vez que pode substituir qualquer nucleotídeo do genoma.
O referido artigo relata a correção de uma dessas mutações (isto é, a T4709M) como exemplo. Esta mutação específica foi selecionada porque existe um modelo de camundongo (RYR1TM/Indel) com essa mutação que desenvolve sintomas claros. Confira o artigo científico no link -> https://www.mdpi.com/2073-4409/13/1/31#
TERAPIA GENÉTICA
A terapia genética é uma grande promessa para o tratamento de doenças genéticas, uma vez que aborda diretamente a raiz do problema. Ao corrigir mutações, a terapia genética tem o potencial de curar milhares de doenças hereditárias.
A descoberta do CRISPR/Cas9 em 2012 foi um marco no desenvolvimento de terapias genéticas. O Crispr/Cas9 é uma espécie de "tesoura genética", que permite à ciência mudar parte do código genético de uma célula. Com essa "tesoura", é possível, por exemplo, "cortar" uma parte específica do DNA, fazendo com que a célula produza ou não determinadas proteínas.
Este sistema usa uma nuclease Cas9 que induz uma quebra da fita dupla do DNA em um local preciso do genoma. Cas9 é direcionado para a sequência do genoma desejada por um único RNA guia (sgRNA). Este sgRNA é um RNA de fita simples complementar a uma sequência de DNA. A proteína Cas9 forma um complexo com o sgRNA e se liga a um motivo adjacente no DNA, induzindo um corte. Após a quebra da cadeia dupla no local do desejado, a célula irá reparar este corte por Reparação Dirigida por Homologia (HDR) se for fornecida uma sequência doadora. No entanto, a percentagem de correção de uma mutação precisa de nucleótidos por HDR é demasiado baixa para ser utilizada no tratamento de doenças hereditárias in vivo . Se nenhuma sequência doadora for fornecida, a célula reparará o corte por junção final não homóloga (NHEJ) e produzirá indels. InDels (inserções e deleções) são adições ou perdas de uma ou mais bases consecutivas na sequência do DNA.
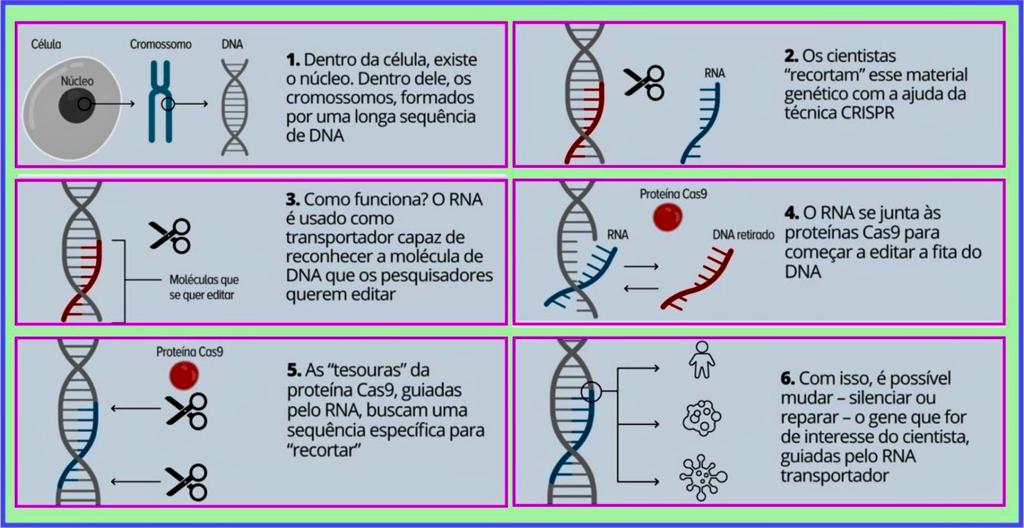
CRISPR/Cas9
Em outubro de 2019, o grupo de David R. Liu publicou uma técnica notável chamada PRIME EDITION. Este sistema pode realizar inserções, deleções direcionadas e todas as 12 conversões de base possíveis.
O Prime Edition ou sistema Edição Prime (em português), é um método de edição de genoma que grava diretamente novas informações genéticas em um local (endereço) de DNA especificado usando uma endonuclease Cas9 prejudicada cataliticamente e fundida com uma transcriptase reversa projetada, programada com um RNA de guia Prime Edition (pegRNA) que especifica o local de destino e codifica a edição desejada. Esta tecnologia realiza modificações no DNA com precisão sem precedentes e oferece vantagens substanciais sobre o sistema tradicional CRISPR/Cas9.
Prime Editing é mais complexo que a edição CRISPR. Ele pode excluir comprimentos longos de DNA causador de doença ou inserir DNA para reparar mutações perigosas, tudo sem desencadear as respostas caóticas (e possivelmente prejudiciais) do genoma introduzidas por outras formas de CRISPR.
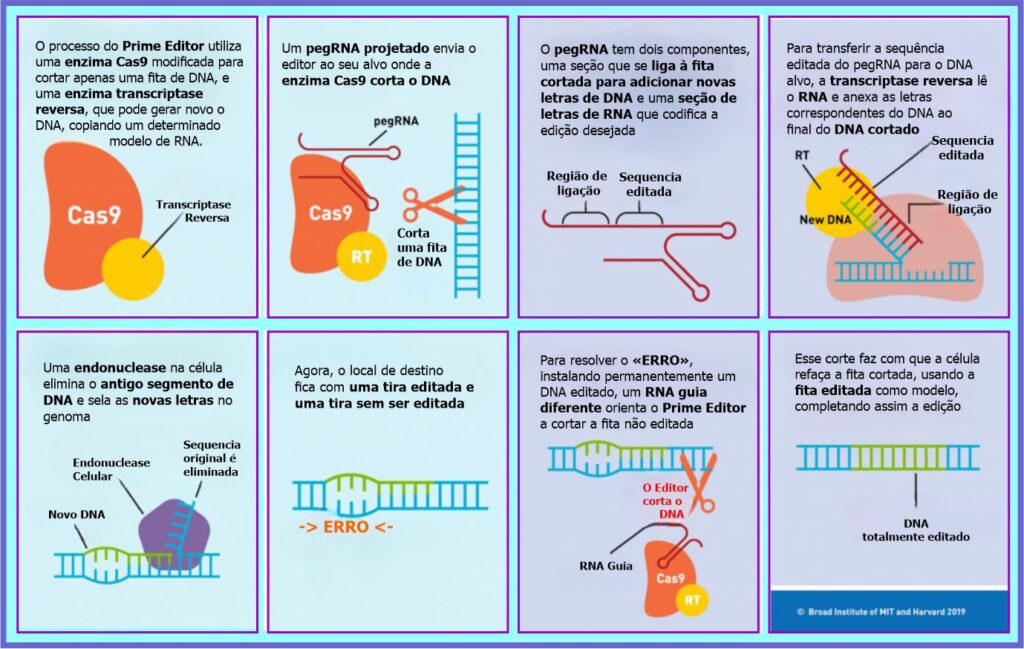
Prime Edition
Em resumo, a técnica CRISPR-Cas9, popularmente utilizada para modificação genética pela comunidade científica, baseia-se na atividade nuclease da enzima Cas9 que corta as duas fitas de DNA, e utiliza a maquinaria de reparo de danos da própria célula. No entanto, o sistema de reparo pode inserir ou deletar letras de DNA, causando efeitos inesperados. Já a nova tecnologia “Prime Editing” ou Edição Prime utiliza uma versão enzima Cas9 que além de reconhecer sequências específicas de DNA, corta apenas uma das fitas da dupla-hélice. Dessa forma, a edição ocorre no local correto do corte através da ação de uma enzima transcriptase e uma fita de RNA guia (pegRNA).
A Miopatia Congênita Centronuclear (MCCN) é uma doença muscular congênita rara caracterizada por fibras celulares com núcleos centralizados proeminentes em biópsias musculares. A doença é clinicamente heterogênea, variando de fenótipos hipotônicos graves já no nascimento até fraqueza muscular leve com início na idade adulta, e pode ter múltiplos modos de herança em associação de causa por mutações nos genes MTM1, DNM2, BIN1 e RYR1.
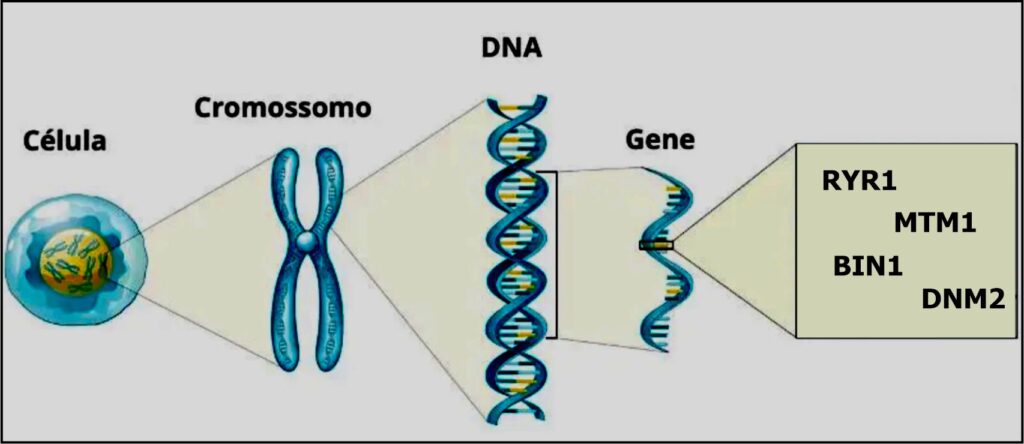
Assim como existe uma grande complexidade no diagnóstico de uma miopatia, tema esse abordado em outra postagem no SORRYR-1.com.br, essas diferentes causas, são também motivo de grande confusão em diagnósticos. É importante consultar um médico especialista para um diagnóstico preciso e assim obter informações detalhadas sobre a mutação específica no caso individual. As diferentes mutações genéticas que causam a Miopatia Congênita Centronuclear podem resultar em variações na gravidade, evolução e sintomas da doença. É importante dizer que a gravidade dos sintomas da doença pode variar de pessoa para pessoa, mesmo com a mesma mutação genética.
Seguem as diferentes causas de origem da Miopatia Congênita Centronuclear (MCCN):
O gene MTM1 é responsável pela codificação de proteína chamada miotubularina. Essa proteína desempenha um papel importante na função muscular, fundamental por atuar como uma enzima fosfatase de desempenho crítico na regulação do tráfego de vesículas dentro das células musculares, particularmente nas fibras musculares esqueléticas. Quando há uma mutação no gene MTM1, a produção ou função da miotubularina é afetada, e sso pode resultar em um acúmulo anormal de vesículas dentro das fibras musculares, levando à fraqueza muscular e outros sintomas associados à Miopatia Congênita Centronuclear.
A dinamina 2, codificada pelo gene DNM2, desempenha um papel crucial na regulação do tráfego de vesículas que transportam proteínas essenciais para a função muscular normal. Ela ajuda a controlar a fusão e divisão dessas vesículas, permitindo a entrega adequada de proteínas contráteis, como a miosina e actina, aos locais onde são necessárias para a contração muscular.
Quando ocorrem mutações no gene DNM2, a função da dinamina 2 pode ser comprometida, resultando em distúrbios que pode levar a fraqueza muscular, em especial nos músculos proximais, e outros sintomas associados a condições da MCCN. Portanto, a dinamina 2 desempenha um papel importante na manutenção da função muscular saudável.
O gene BIN1 codifica a proteína anexina A2, que está envolvida na regulação das membranas celulares e no tráfego de vesículas nas células musculares. Essas funções desempenhadas pela anexina A2 são essenciais para a saúde e a função das fibras musculares, a mutação nesse gene afeta negativamente a estrutura e a função das células musculares, o que resulta em fraqueza muscular e outros sintomas associados à CCNM. A gravidade e a apresentação dos sintomas podem variar com base na mutação específica do gene BIN1 envolvida.
O gene RYR1 codifica o receptor de rianodina 1, que é uma proteína essencial para a função das fibras musculares. O receptor de rianodina está envolvido na liberação (válvula de controle) de cálcio das reservas intracelulares, um processo fundamental para a contração e relaxamento muscular.
As doenças musculares são aquelas que afetam a estrutura e funcionamento do músculo, sendo as principais: as distrofias musculares, as miopatias congênitas, as miopatias inflamatórias e as miopatias endócrinas e metabólicas. É importante destacar que cada uma delas possui suas variações que também se diferenciam.
Essas doenças já foram muito confundidas em diagnósticos no passado, e fico triste, porque isso ainda tem acontecido nos dias de hoje, mesmo com os avanços científicos. A única razão que acredito ser ainda a causa para essa confusão nesses diagnósticos seria por essas doenças serem consideradas “doenças raras”, portanto, muitas vezes desconhecidas por parte da comunidade médica. Assim, pode haver a falha no momento dos exames clínicos, ponto inicial para diagnóstico de qualquer doença.
Eu mesmo vivi uma experiência dessa, pois no decorrer de grande parte da minha vida eu recebi vários “diagnósticos” de Distrofia Muscular Congênita (DMC) do tipo: Duchenne, Facioescapuloumeral e Cinturas. E, os prognósticos foram do pior a até o mais brando. Estes diagnósticos ou hipóteses de diagnósticos vieram até de importantes instituições, como de uma clínica indicada pelo MDA (Muscular Distrophy Association), maior referência ligada a essa doença.
Deve-se levar em consideração que, naquela época, pouco se sabia sobre essa doença, nem tão pouco sobre a genética humana; contudo, um erro de diagnóstico hoje em dia seria inaceitável. Essa situação causou em mim grandes transtornos, de emocional aos físicos. Somente aos 44 anos de idade foi que finalmente obtive meu correto e “definitivo diagnóstico”, ou seja, de que sou portador de Miopatia Congênita Centronuclear (MCC), causada pela mutação no gene RYR1.
A Distrofia Muscular Congênita e a Miopatia Congênita Centronuclear apresentam várias características em comum, tais como: são doenças de origem genética, afetam os músculos esqueléticos, caracterizam-se clinicamente por hipotonia e fraqueza muscular, geralmente apresentam-se desde o nascimento, têm curso clínico estático ou lentamente progressivo. Essas doenças não tem cura, e o tratamento envolve terapia de suporte, como fisioterapia, dispositivos de mobilidade e, em alguns casos, medicamentos. Mesmo assim, as duas doenças neuromusculares diferem entre si.
Daí, eu volto com a questão sobre as falhas nos diagnósticos, já que muitos médicos se prendem somente ao resultado do exame genético e não conhecerem os sinais clínicos das diferentes doenças e particularidades dos indivíduos afetados.
Assim, essas noções devem ser levadas em consideração por três razões: Primeiro, muitas das miopatias congênitas podem ser causadas por mutações em mais de um gene, o que sugere um impacto da heterogeneidade genética. Segundo, mutações no mesmo gene podem causar diferentes patologias musculares. Terceiro, a mesma mutação genética pode levar a diferentes características patológicas e sintomatológicas em membros da mesma família ou no mesmo indivíduo em idades diferentes.
Em resumo, eu destacaria que tanto a Distrofia Muscular, quanto a Miopatia Congênita Centronuclear são de origem genética, mas distintas em termos de suas características clínicas e podem variar em gravidade de pessoa para pessoa. Enquanto a Distrofia Muscular envolve a degeneração progressiva dos músculos devido a problemas na estrutura das proteínas musculares, a Miopatia Congênita Centronuclear é caracterizada por uma anormalidade na localização dos núcleos das células musculares. Assim, é importante consultar um médico especialista para um diagnóstico preciso, para que se possa ser feito um acompanhamento adequado do caso, pois o tratamento pode variar dependendo da condição clínica específica de cada indivíduo.
OS INDIVIDUOS PORTADORES DE DOENÇAS RELACIONADAS AO RYR-1 DEVEM ESTAR SEMPRE ATENTOS ÀS EVENTUAIS E POSSÍVEIS COMPLICAÇÕES CAUSADAS PELA DOENÇA
Como já dito anteriormente, não existe um tratamento específico para as doenças relacionadas ao RYR-1, e toda conduta médica diante do indivíduo afetado depende da etiologia, e sintomatologia de manifestação da doença, tendo sempre em vista as possíveis e eventuais complicações causadas pela doença. A morbidade e mortalidade de pessoas afetadas por miopatias relacionadas ao RYR-1 estão diretamente relacionadas à gravidade da doença, bem como à presença de outras comorbidades. As complicações mais comuns das miopatias relacionadas ao RYR-1 são as com questões respiratórias, ortopédicas, dentre outras.
Complicações Respiratórias
Dentre os diversos sistemas que temos em nosso corpo e que faz o organismo humano estar em funcionamento, destaca-se o sistema respiratório, para todos, de importância vital. Seu funcionamento depende diretamente de alguns músculos, dentre eles, dois diafragmas (músculos entre os pulmões e o abdome), os músculos entre as costelas, e os músculos no pescoço e garganta. As doenças neuromuscular podem causar a fraqueza destes músculos, os fazendo cansados e fatigados, difícultando assim a inspiração e expiração de maneira efeitiva. O funcionamento respiratório ineficaz pode abaixar a oxigenação e elevar os níveis de gás carbônico no seu sangue, fazendo assim com que os músculos fiquem ainda mais fracos, criando assim um ciclo vicioso,totalmente nocivo ao doente.
Assim como dentre outros sintomas, as complicações causadas pelas doenças relacionadas ao RYR-1, as respiratórias estão entre as mais comuns, e podem variar conforme a gravidade da doença. Alguns indivíduos podem não ter problema algum, outros podem ter problemas leves, mas que precisam de ajuda para respirar enquanto dormem ou quando estão doentes, e em casos graves, podem precisar de um ventilador para ajudá-los a respirar durante todo tempo. Portanto, dependendo do acometimento da doença no indivíduo, os médicos costumam fazer o acompanhamento do sistema respiratório regularmente.
O teste de função pulmonar (TFP) é uma forma de o pneumologista avaliar a presença, tipo e gravidade dos problemas respiratórios de alguém.
A polissonografia, ou “estudo do sono”, é feito para identificar, diagnosticar e tratar distúrbios do sono, causados por complicações no relaxamento dos músculos da garganta, situação esta, comum nos indivíduos afetados pelas miopatias causadas pelo RYR-1. O exame é feito durante o sono, totalmente indolor e não invasivo, e pode ser capaz de identificar por exemplo, a apneia obstrutiva do sono (obstrução temporária ou completa da respiração durante o sono) ou insuficiência respiratória (incapacidade de inalar oxigênio suficiente e exalar dióxido de carbono suficiente). Outra abordagem médica visando a prevenção e administração de complicações de problemas respiratórios associados à doenças causadas pelo RYR-1, são as imunizações com vacinas contra gripe, pneumonia, e qualquer outro virus que pode afetar o sistema respiratório, e a fisioterapia visando ajudar na tosse, com técnicas para facilitar a limpeza do muco, assim como para o tratamento da apneia do sono.
Complicações Ortopédicas
As complicações ortopédicas de natureza das doenças neuromusculares são bastantes comuns, tanto por desenvolvimento secundário ao desequilíbrio muscular, como por causa de trauma, distúrbios do nascimento, ou de caráter degenerativo. As complicações ortopédicas mais comuns entre pessoas afetadas por uma doença relacionada ao RYR-1 são: fraqueza, contraturas, luxações do quadril, escoliose, e outras deformidades da coluna vertebral.
A contratura muscular ocorre quando o músculo se contrai de maneira incorreta, seja por este estar muito fraco ou muito curto, não voltando assim ao seu estado normal de relaxamento. A contratura pode levar uma articulação não ter toda a sua amplitude de movimento, a tornando fixa ou rígida, promovendo a substituição do músculo por tecido conjuntivo rígido, ocasionando assim problemas com a cápsula articular, até deformidades físicas. O especialista médico, no caso o ortopedista, precisa determinar a causa da contratura para assim determinar a melhor forma de tratá-la, que pode ser através da fisioterapia, e utilização de órteses.
Luxação do quadril significa que o osso do quadril (fêmur) se posiciona de fora de sua junta ou fora de seu encaixe. Na maioria dos casos, o deslocamento ou luxação do quadril não interfere na capacidade de andar da pessoa. Não existe nenhuma regra padrão no tratamento de luxações do quadril em pessoas com doenças neuromusculares, e esta abordagem dependerá da idade do paciente, se existe controle e força muscular suficiente para apoiar o quadril, se o deslocamento afeta um ou ambos os lados, se o deslocamento afeta o dia a dia do indivíduo, e lhe cause sintomas como a dor. As luxações de quadril podem ser tratadas com a utilização de tala ou dispositivo semelhante. Contudo, caso isso não consiga controlar o deslocamento, especialistas poderão considerar a cirurgia.
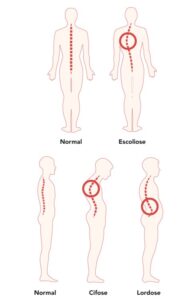
As deformidades na coluna vertebral mais comuns em pacientes afetados por uma miopatia relacionada ao RYR-1, é a Escoliose, que se trata de uma curva lateral da coluna vertebral, a Cifose que é a coluna curva muito para trás (“corcunda”), e a Lordose, que é a coluna curva muito para frente (“arqueada voltar"). Essas deformidades, têm como causa a fraqueza muscular, e deficiência de mobilidade, e podem piorar com a idade do indivíduo. As deformidades da coluna vertebral podem fazer com que a parede torácica se torne estreita ao ponto de interferir no funcionamento dos pulmões e diafragma, no equilíbrio, capacidade de movimentação dos braços, assim como na qualidade de vida do indivíduo. Um importante ponto de atenção e preocupação para o indivíduo afetado por uma miopatia causada pelo RYR-1, dado à fraqueza dos músculos respiratórios, é a Síndrome da Insuficiência Torácica, que é uma condição em que o tórax não consegue suportar a respiração ou crescimento do pulmão. No caso do paciente apresentar qualquer sinal de deformidade na coluna vertebral, é muito importante o acompanhamento vigilante de um ortopedista ou neurologista, que buscará o seu não agravamento, através da indicação de um trabalho de reeducação postural, mas sobretudo a fisioterapia funcional e respiratória.
Indivíduos portadores de doença relacionada com RYR-1, além de complicações respiratórias, e ortopédicas, podem também desenvolver problemas com músculos oculares, mastigação, deglutição e respiração, fraqueza muscular podendo ser de consequências leve a severa, a até a uma potencial reação fatal a certas formas de anestesia conhecidas como Hipertermia Maligna. A Hipertermia Maligna ocorre em indivíduos suscetíveis a este tipo de reação, como em aqueles afetados por mutação no RYR-1, e acontece como uma resposta do corpo a certos gases anestésicos e a um certo tipo de relaxante muscular usado para bloquear a sensação de dor, utilizado durante um procedimentos de anestesias em salas de cirurgia, salas de emergência e unidades de terapia intensiva (UTI). A Hipertermia Maligna é uma reação do corpo em que pode apresentar rigidez muscular, deterioração das fibras musculares (rabdomiólise), superaquecimento do corpo (febre alta), aumento dos níveis de ácido no sangue e outros tecidos (acidose), aumento da frequência cardíaca, e se o paciente não for tratado a tempo, HM pode resultar em insuficiência renal, cérebro danos, parada cardíaca, falência de órgãos adicionais e até morte. Existem atualmente algumas drogas específicas que são conhecidas por desencadear a Hipertermia Maligna, que são: (halotano, enflurano, isoflurano, sevoflurano e desflurano) e, provavelmente, succinilcolina. A sugestão preventiva que se dá a um indivíduo suscetível a esta reação, é comunicar aos seus familiares, pessoas de sua convivência, plano de saúde, além de fixar avisos em seus documentos pessoais, aparelho de celular, com a informação de advertência sobre o risco eminente da Hipertermia Maligna, destacando o nome das drogas a serem evitadas em caso de emergência.

