O gene RYR1, descrito em 1989 por MacLennan é responsável por dar as instruções para a proteína do receptor de rianodina 1 (RYR1), que é um canal de cálcio crucial para a contração e relaxamento dos músculos esqueléticos.1 A função principal do gene é codificar o canal que libera íons de cálcio do retículo sarcoplasmático (um reservatório dentro da célula muscular) quando o músculo é ativado por um sinal elétrico. Quando o canal RYR1 se abre, o cálcio entra no citoplasma, permitindo que os músculos se contraiam para permitir o movimento. Variantes (mutações) neste gene podem levar a doenças musculares como miopatias congênitas: Doença do Núcleo Central (Central Core), Doença Multi-Minicore, Miopatia Centronuclear e Desproporção Congênita do Tipo de Fibra. O estudo destas doenças permitiu o sequenciamento do gene RYR1 e a descoberta da relação de variantes neste gene e manifestações clínicas como Hipertermia Maligna (HM), Rabdomiólise e Rabdomiólise por Esforço (RE).
RYR1 está localizado no cromossomo 19q13.1 e codifica o Receptor de Rianodina Tipo 1 (RYR1). O gene RYR1 compreende 106 exons e codifica uma grande proteína com 5038 aminoácidos. Devido ao seu tamanho e complexidade consideráveis, o RYR1 tem sido historicamente um gene desafiador para estudo. Durante muitos anos, os esforços de pesquisa concentraram-se principalmente em três domínios (N-terminal, central e C-terminal) considerados pontos críticos de mutação. No entanto, com o advento das tecnologias de Sequenciamento de Nova Geração (NGS), tornou-se possível analisar o gene inteiro. Como resultado, inúmeras novas variantes foram identificadas fora dos pontos críticos previamente conhecidos. Até o momento, mais de 1000 variantes distintas do gene RYR1 foram relatadas (HGMD; https://www.hgmd.cf.ac.uk/ (acessado em 04 de dezembro de 2025) e LOVD; https://databases.lovd.nl/shared/genes/RYR1 (acessado em 04 de dezembro de 2025), porém apenas 72 foram classificadas como variantes diagnósticas pelo Grupo Europeu de Hipertermia Maligna (EMHG; https://www.emhg.org/diagnostic-mutations (acessado em 04 de dezembro de 2025)). A grande maioria dessas variantes são de sentido trocado, enquanto inserções, deleções e duplicações são relativamente incomuns, representando menos de 10% de todas as variantes patogênicas conhecidas no gene RYR1.
A Hipertermia Maligna (HM) é uma síndrome hipermetabólica farmacogenética que se manifesta como uma crise aguda após a exposição de indivíduos suscetíveis a agentes halogenados e/ou succinilcolina. A crise aguda pode se manifestar clinicamente com hipertermia, hipercapnia, taquipneia, taquicardia, acidose metabólica, hipercalemia, rigidez muscular e rabdomiólise, sendo uma condição extremamente grave. Ocorre em pacientes de todas as etnias e distribuições geográficas, sendo duas vezes mais comum nos homens e menores de 19 anos de idade (50% dos casos). Se não tratada adequadamente, óbito ocorre em 80%–90% dos pacientes. O tratamento consiste em evitar as crises e abordagem seguindo protocolos rígidos nas crises agudas, incluindo-se uso de dantrolene de sódio (reduz a liberação de cálcio do retículo sarcoplasmático do músculo estriado esquelético por limitar a ativação do receptor de rianodina RYR1).2,3
A prevalência da crise de HM é variável, de 1:10.000 em crianças a 1:50.000 em adultos. A frequência relacionada a procedimentos anestésicos varia de 1 em 10.000 a 1 em 250.000, dependendo da população e dos critérios diagnósticos. A suscetibilidade à HM tem sido associada a genes ligados ao metabolismo do cálcio e que codificam proteínas do complexo de acoplamento excitação-contração do músculo esquelético, com uma frequência de 1:217–1:2750 na população geral. O principal gene implicado na suscetibilidade à HM é o RYR1, responsável por aproximadamente 75% dos casos geneticamente confirmados. Famílias raras apresentam variantes nos genes CACNA1S (1%), STAC3 (<1%) ou ASPH.
A apresentação clínica entre portadores de variantes do RYR1 é altamente variável, e o panorama genético é marcado por alta heterogeneidade alélica, penetrância incompleta e expressividade variável. Variantes patogênicas nesse gene resultam em liberação desregulada de cálcio do retículo sarcoplasmático, levando à contração muscular sustentada e crise metabólica.
Devido à heterogeneidade genética e à possibilidade de herança poligênica, o padrão ouro no diagnóstico de suscetibilidade à HM é o teste de contratilidade in vitro fenotípica (IVCT) para o grupo europeu ou o teste de contratilidade com cafeína-halotano (CHCT) para o grupo norte-americano. Atualmente, apenas um teste de contratilidade negativo pode excluir a suscetibilidade à HM, enquanto um teste genético negativo não o faz. No entanto, esforços têm sido feitos para melhorar a capacidade de detecção do teste molecular, que poderia ser uma ferramenta diagnóstica menos invasiva. Por outro lado, variantes no gene RYR1 são muito frequentes na população, e a identificação e classificação dessas variantes como patogênicas requerem testes adicionais e curadoria. Apenas 72 variantes patogênicas ou provavelmente patogênicas no RYR1 são reconhecidas de acordo com a lista do Grupo Europeu de Hipertermia Maligna (EMHG) ( https://www.emhg.org/genetic-scoring-matrix ; acessado em 18 de agosto de 2025), e, além dos critérios do EMHG, existem os critérios do painel de especialistas em curadoria de variantes do ClinGen MHS (VCEP). Variantes missense são as mais comuns, enquanto inserções e duplicações representam menos de 10%. Os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína.
Cada país ou região apresenta diferenças no tipo e na frequência de variantes associadas à HM. A frequência de variantes relatadas em pacientes com HM varia de 37% a 87,5%, sendo as variantes RYR1 p.R614 e p.G2434R as mais frequentes. No entanto, não existem dados genéticos populacionais da América do Sul, exceto pela descrição de casos isolados e famílias. Em estudo recente, desenvolvido na Unifesp – Escola Paulista de Medicina, foram revisados os dados clínicos e laboratoriais de todas as famílias encaminhadas para avaliação na Unidade Brasileira de HM devido a histórico pessoal ou familiar de HM durante anestesia. Foram coletados dados demográficos e clínicos, bem como níveis séricos de creatina quinase (CK), resultados do teste de contratilidade in vitro (TCIV) e resultados de estudos anatomopatológicos do músculo esquelético. A análise molecular foi realizada por meio de sequenciamento de exoma completo (NGS). Pacientes com e sem variantes foram comparados. Variantes no gene RYR1 foram encontradas em 38 pacientes (62,2%), e nenhuma variante foi identificada em 20 pacientes (32,7%). Mais de uma variante no RYR1 foi encontrada em seis indivíduos. Variantes no gene CACNA1S foram encontradas em três pacientes (4,9%), todos com variantes concomitantes no RYR1. Três pacientes apresentaram variantes no gene STAC3 (4,9%). Comparando os grupos de pacientes com variantes no RYR1 com o grupo sem variantes nesse gene, observou-se que o primeiro grupo apresentou valores séricos mais elevados de CK, maior frequência de ptose, estrabismo, e maior amplitude de contratura no TCIV após a administração de cafeína ou halotano. Nesta avaliação preliminar de indivíduos brasileiros com histórico de hipertermia maligna, a frequência de variantes no RYR1 foi semelhante à de relatos anteriores em outros países, porém houve maior frequência de variantes nos genes STAC3 e CACNA1S.4
Também em estudo da Unifesp, foi identificada uma variante rara: duplicação no gene RYR1 na variabilidade do fenótipo de susceptibilidade à Hipertermia Maligna, em uma família com dois irmãos afetados portadores de uma inserção de 18 pares de bases no éxon 91 do gene RYR1, resultando em uma duplicação em fase de 6 aminoácidos (c.12835_12852 dupGAGGGCGCGGCGGGGCTC: 162 p.G4279_T4284insAAGLEG). A expressão relativa do mRNA do gene RYR1 no músculo dos dois pacientes identificou uma redução de aproximadamente 50%, sugerindo um possível alelo hipomórfico. Este achado levanta a questão que os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína. Este estudo reforça a possibilidade de que a duplicação nessa região possa causar defeitos estruturais e um fenótipo mais grave nos pacientes.5
A Rabdomiólise é uma condição potencialmente fatal que envolve a rápida dissolução do músculo esquelético em resposta a uma variedade de fatores desencadeantes, clinicamente caracterizada por um aumento súbito e acentuado, seguido de uma queda nos valores séricos de creatina quinase (CK) As causas mais comuns de rabdomiólise são lesões por esmagamento secundárias a traumas, esforço físico extremo e miopatias metabólicas. As principais características incluem dor muscular e uma elevação súbita e transitória dos valores séricos de CK. A rabdomiólise grave é frequentemente acompanhada por aumento da excreção urinária de mioglobina (mioglobinúria), o que pode levar à insuficiência renal aguda (IRA) e a uma crise metabólica potencialmente fatal. A ampla gama de complicações (por exemplo, insuficiência renal aguda, arritmias cardíacas, síndrome compartimental, coagulação intravascular disseminada) enfatiza a relevância clínica da rabdomiólise em diversas especialidades médicas.
Estudos de coorte retrospectivos focados em rabdomiólise em pacientes hospitalizados na era pré-sequenciamento de nova geração (NGS) concentraram-se particularmente em fatores desencadeantes externos como a principal causa de um evento de rabdomiólise. Esses estudos identificaram toxinas exógenas (drogas ilícitas, álcool), trauma muscular direto, infecções e exercícios extenuantes (Rabdomiólise por Esforço) como alguns dos fatores desencadeantes mais comuns. Exames genéticos de última geração têm associado mais de 30 genes a uma maior suscetibilidade à rabdomiólise. Contudo, um desafio fundamental na abordagem diagnóstica reside na consideração de quais pacientes necessitam de triagem genética diagnóstica após um episódio de rabdomiólise para identificar uma doença neuromuscular ou metabólica pauci-sintomática ou assintomática. Dentre os genes relacionados, destaca-se o RYR1 e variantes nele podem ser responsáveis por uma proporção substancial de pacientes que apresentam sintomas inexplicáveis de rabdomiólise e/ou mialgia por esforço.6
Com o intuito de revisar a abordagem diagnóstica genética da rabdomiólise, uma pesquisa na Holanda foi realizada, tendo como palavra chave o acrônimo 'RHABDO': Recurrent episodes (episódios recorrentes); HyperCKaemia persisting 8 weeks after the event (aumento de CK persistente após 8 semanas do evento); Accustomed exercise—the intensity of the exercise cannot sufficiently explain the rhabdomyolysis event (exercício de costume – a intensidade do exercício não explica por si o evento de rabdomiólise); Blood CK > 50× the upper limit of normal (ULN) or >10,000 IU/L in female Caucasian patients (aumento de CK > 50x ou > 10 000 UI/L em mulheres); Drugs/medication and other exogenous triggers are insufficient to explain the event (drogas/medicações e outros agentes exógenos são insuficientes para explicarem o evento); and Other affected family members or other exertional symptoms (e.g., severe muscle cramps or swelling) (outros familiares afetados ou com sintomas relacionados ao exercício físico). O acrônimo foi baseado em uma revisão da literatura e em nossa experiência clínica e, portanto, atingiu o nível de evidência de opinião de especialistas. A relevância do RHABDO é ainda mais reforçada por um recente workshop do Centro Neuromuscular Europeu (ENMC) envolvendo 21 médicos e pesquisadores de 12 países diferentes, que enfatizou a necessidade de pesquisa coordenada nesta área. Neste estudo retrospectivo bicêntrico, 122 pacientes foram incluídos. Os fatores desencadeantes mais frequentemente relatados que contribuíram para eventos de rabdomiólise foram exercício (72%), febre/infecção (22%) e/ou medicação (18%). Eles foram submetidos a avaliação genética através de painéis genéticos relacionados à miopatia metabólica (82%), sequenciamento de Sanger (49%) e sequenciamento de exoma completo (NGS) (24%), dos quais 52 pacientes (43%) foram submetidos a múltiplos métodos. Uma variante (provavelmente) patogênica foi identificada em 13 pacientes (11%), todos com ≥2 características de RHABDO presentes. O valor preditivo positivo para ≥2 características foi de 14%, enquanto o valor preditivo negativo foi de 100%. Variantes no gene RYR1 foram descritas em quatro pacientes, um relacionado com esforço (RE).7
A Rabdomiólise por Esforço (RE) é uma degradação muscular patológica associada à atividade física extenuante e agravada por múltiplos fatores de risco. Estes incluem baixo nível de condicionamento físico, alto índice de massa corporal, infecção viral em curso e altitude e temperatura elevadas. A sua incidência é de aproximadamente 36,5 por 100.000 pacientes-ano em atletas ou uma taxa semelhante em militares. De modo geral, as diferenças fundamentais entre a RE e outras formas de rabdomiólise residem em suas causas, fatores desencadeantes e populações afetadas. A patologia celular da lesão por esforço repetitivo (LER) centra-se na ruptura da integridade das células musculares, particularmente no que diz respeito à homeostase iônica e à produção de energia. Exercícios extenuantes ou incomuns podem causar lesão direta ao sarcolema e/ou levar à falha na produção de energia, comprometendo a função de bombas iônicas essenciais, como a Na + /K + -ATPase e a Ca2 + -ATPase. Esse comprometimento aumenta a permeabilidade celular aos íons sódio, resultando em um influxo significativo de cálcio (Ca2 +) para as fibras musculares. O aumento da concentração intracelular de cálcio ativa enzimas dependentes de cálcio, incluindo proteases e fosfolipases, que iniciam a destruição de proteínas miofibrilares, citoesqueléticas e de membrana. Esse processo leva à necrose das fibras musculares, liberando conteúdos intracelulares como CK, mioglobina e eletrólitos no fluido extracelular e na circulação sanguínea. O ciclo vicioso resultante envolve contração muscular sustentada devido ao aumento do cálcio, o que esgota ainda mais as reservas de energia e exacerba o dano muscular.
Embora a via celular geral envolvendo sobrecarga de cálcio e depleção de energia, seja compreendida como o mecanismo de dano às células musculares, os mecanismos subjacentes específicos da rabdomiólise (RB) não são universalmente compreendidos. Estudos em modelos animais, como cavalos suscetíveis à RB recorrente, utilizaram com sucesso a análise do transcriptoma (RNA-seq ou análise de microarray) para revelar alterações na expressão gênica em vias relacionadas à regulação do cálcio, estresse oxidativo e função mitocondrial, demonstrando que essas alterações moleculares podem persistir mesmo entre os episódios. No entanto, as fontes fornecidas não oferecem uma visão abrangente de pesquisas semelhantes em larga escala sobre o transcriptoma, conduzidas especificamente em populações humanas com RB. Um estudo elegante com sequenciamento de RNA em amostras de músculo esquelético de 19 pacientes humanos com histórico de RE, coletadas no mínimo seis meses após o evento de RE mais recente, e oito controles saudáveis para investigar o perfil transcriptômico da RE revelou uma forte supressão da função mitocondrial. Essa supressão incluiu as vias da “cadeia de transporte aeróbico de elétrons” e da “fosforilação oxidativa”, indicando comprometimento da produção de energia. Por outro lado, houve uma regulação positiva de genes associados à adesão e às vias relacionadas à matriz extracelular (aumento do desenvolvimento da matriz extracelular), indicando restauração ativa da função muscular em casos de RE meses após o evento agudo.8
Em síntese, destacamos o que estas pesquisas nos ensinam:
Como o gene RYR1 afeta a função muscular
Associação de variantes (mutações) no RYR1 e Hipertermia Maligna (HM)
Como o defeito no RYR1 leva à Rabdomiólise
O que acontece durante a Rabdomiólise por Esforço (RE)
O que os indivíduos com variantes no RYR1 precisam saber
Hipótese
Referências Bibliográficas:
A Miopatia Congênita Centronuclear é uma doença que causa muitas exclamações, mas também, interrogações. Sempre que as pessoas chegam até mim para questionar sobre minha doença, elas fazem uma exclamação dizendo, “nunca tinha ouvido sobre sua doença !”, e em seguida questionam como ela me afeta, e mais uma vez elas exclamam dizendo, "puxa vida !", e por ultimo exclamam novamente, “essa é uma super rara !”, daí respondo, "sim, no pé da palavra, ela é uma Doença Rara ou talvez uma Doença Ultrarrara.
A Miopatia Congênita Centronuclear decorrente de mutações no gene RYR1 é classificada como Doenças Raras ou Doenças Ultrarraras ?
Entender essas condições é essencial para oferecer o suporte adequado e assegurar abordagens terapêuticas personalizadas tanto para o paciente quanto para seus familiares. Doenças raras nem sempre recebem a atenção que merecem porque afetam relativamente poucas pessoas. Muitas vezes, pode levar anos para que uma pessoa receba o diagnóstico correto de uma doença rara, e cerca de 95% das doenças raras e ultrarraras ainda não têm tratamento.
Diferentes partes do mundo, as doenças raras e ultrarraras são identificadas e tratadas de maneiras diferentes, mas de um modo genérico a definição é mais ou menos assim:
Uma Doença Rara é aquela que acomete no máximo 65 pessoas a cada 100.000 habitantes, o que corresponde a uma prevalência aproximada de 1 para cada 1.500 indivíduos. A maior parte dessas condições tem origem genética, embora algumas possam surgir devido a fatores infecciosos ou relacionados ao sistema imunológico.
A Doença Ultrarrara, como o próprio nome indica, corresponde a condições ainda mais incomuns, com uma incidência de cerca de 1 caso para cada 50.000 indivíduos. Por serem extremamente infrequentes e pouco conhecidas, essas doenças geralmente apresentam maior dificuldade diagnóstica e exigem cuidados altamente especializados e personalizados.
Com relação à questão levantada no início do texto, a prevalência exata da Miopatia Congênita Centronuclear causada especificamente pela mutação no gene RYR-1 é desconhecida.
A dificuldade em determinar um número exato ocorre porque:
• Ela é uma doença ultrarrara por estar dentro de um grupo de doenças já raras, as Miopatias Congênitas.
• Os dados epidemiológicos costumam ser apresentados para a Miopatia Congênita Centronuclear (MCCN) considerando o conjunto das patologias que compõem esse grupo, que inclui mutações nos genes MTM1, DNM2, BIN1 e RYR1.
• O gene RYR1 está associado a um espectro de doenças musculares conhecidas como Doenças Relacionadas ao RYR1 ou RYR1-DR, sendo a forma Centronuclear apenas uma das possíveis manifestações, juntamente com a Miopatia Central Core (MCC), e outras.
Embora não haja um número específico de prevalência para a Miopatia Congênita Centronuclear causada pela mutação no RYR1, podemos contextualizar com base nas informações disponíveis para as condições relacionadas:
• No grupo da Miopatia Congênita Centronuclear (MCCN) em geral, a prevalência geral do grupo é desconhecida. A forma mais comum dentro deste grupo é a Miopatia Miotubular (ligada ao X) causada por mutações no gene MTM1, cuja incidência é estimada em cerca de 1 em 50.000 indivíduos do sexo masculino.
• No grupo das Doenças Relacionadas ao RYR1 (RYR1-DR) em geral, a doença mais frequentemente associado é a Miopatia Central Core (MCC), que é a forma mais comum de miopatia congênita não distrófica.
Um estudo de uma série pediátrica na Espanha estimou a incidência de Miopatias Relacionadas ao RYR1, que abrange a Centronuclear e a Central Core, se mostra em cerca de 1 em 10.000 nascidos vivos na área de estudo.
Em síntese, a mutação no gene RYR1 está associada a um fenótipo de ocorrência rara. A prevalência específica do subtipo Centronuclear (MCCN) ainda não foi estabelecida de forma isolada na literatura médica, sendo considerada extremamente baixa. Entretanto, não há consenso atual sobre classificá-la como no grupo de Doenças Raras ou no grupo de Doenças Ultrarraras.
O estilo de vida levado por um indivíduo afetado por uma miopatia congênita é fundamental para o controle dos sintomas da condição, embora não existam tratamentos específicos para a cura da doença. Essas intervenções são consideradas tratamentos de suporte e visam preservar a função muscular, maximizar a independência, e melhorar a qualidade de vida do paciente.
O tratamento da miopatia congênita deve ter uma abordagem multidisciplinar e individualizada, focada na reabilitação e no suporte contínuo para otimizar a funcionalidade e o bem-estar. Esse estilo de vida a que me refiro, seria sustentado por um tripé de condutas, sintetizado no Controle de Stress, Nutrição, e Atividade Física.
 Controle do Stress
Controle do Stress
O controle do stress (stress management) é de extrema importância para a manutenção da saúde e qualidade de vida de indivíduos portadores de uma miopatia congênita. Embora não haja a cura ou tratamento específico para a doença em si, muitas vezes a única terapia indicada é de suporte, como fisioterapia.
Contudo, existem outras condutas que podem ser tomadas visando a mitigação dos sintomas da doença, melhoria na qualidade de vida dos indivíduos, e que inclusive interferem positivamente no prognóstico da doença. Essa conduta diz respeito a uma série de mudança ou incorporação de hábitos vida, chamada de Controle de Stress, e que é composta em vários fatores interligados:
 Nutrição
Nutrição
A qualidade de vida de um indivíduo afetado por uma miopatia é fortemente influenciada pela capacidade de manter a funcionalidade e a autonomia o máximo possível, apesar da fraqueza muscular, que é o sintoma mais comum. Neste contexto, a importância do controle nutricional é crítica e multifatorial, sendo uma parte essencial para seu tratamento de suporte.
No caso específico da Miopatia Centronuclear, por ser uma doença neuromuscular que causa fraqueza muscular significativa (hipotonia), pode levar a várias complicações que exigem um acompanhamento nutricional individualizado e rigoroso, fundamental para minimizar a perda de massa muscular, garantir o aporte energético adequado, e prevenir complicações secundárias à fraqueza. Esse manejo nutricional não visa curar a doença, mas sim otimizar a saúde geral, apoiar a função muscular e respiratória, e prevenir complicações que impactam severamente a qualidade de vida.
Não existe uma dieta ou protocolo de suplementação único para todos os indivíduos com miopatia congênita. A dieta e suplementação deve garantir um aporte energético suficiente para as necessidades metabólicas e para preservar a já tão sofrida massa muscular. O cálculo do gasto energético total precisa ser individualizado, considerando o nível de atividade física e a gravidade da miopatia.
O plano deve ser altamente individualizado e baseado na avaliação clínica, estado nutricional atual e exames laboratoriais. Para garantir segurança e eficácia, as intervenções nutricionais e a suplementação devem ser sempre orientadas por um médico e um nutricionista especializados ou com conhecimento em doenças neuromusculares. Esse planejamento nutricional se baseia em algumas premissas básicas:
Em resumo, uma nutrição bem planejada e a suplementação direcionada são ferramentas de suporte poderosas que visam otimizar a função muscular residual, prevenir deficiências nutricionais, apoiar a saúde óssea e melhorar a qualidade de vida do indivíduo com miopatia congênita.
 Atividade Física
Atividade Física
Sempre digo que a atividade física, no contexto das miopatias congênitas, vai além da função de mero suporte clínico, pois ela se estabelece como um elemento fundamental na promoção da qualidade de vida. A atividade física é crucial não apenas para retardar o declínio funcional, mas também para promover o bem-estar psicossocial do indivíduo afetado pela doença.
Para indivíduos com miopatia congênita, a atividade física é uma ferramenta terapêutica que busca promover a capacidade do indivíduo agir de forma mais autônoma possível, além de atuar na conexão social e dignidade pessoal, situações estas tão importantes quanto qualquer intervenção médica para alcançar uma qualidade de vida.
A atividade física capacita o indivíduo, dando-lhe um sentimento de propósito, controle e autoestima, que são um pilares da qualidade de vida.
A Declaração de Consenso sobre Padrão de Cuidados para Miopatias Congênitas, publicada em 2012 no Journal of Child Neurology, tratou a atividade física no contexto de se enfatizar a importância de um programa estruturado, mas altamente individualizado, para manter a função e a mobilidade.
O consenso reconheceu que, embora não exista cura, a fisioterapia e a terapia ocupacional (TO) são cruciais para o manejo da doença, e como promoção da qualidade de vida do indivíduo.
As diretrizes do consenso de 2012 sobre atividade física e reabilitação se referiam principalmente aos seguintes pontos:
Em essência, a declaração de consenso enfatizou que a atividade física para miopatias congênitas não é uma atividade de "ganho de força" típica, mas sim uma intervenção de manutenção e reabilitação focada em preservar a função e evitar as complicações secundárias da fraqueza, mas sobretudo e principalmente como agente de promoção de qualidade de vida.
ARMGO PHARMA PÚBLICA IMPORTANTES E POSITIVOS RESULTADOS DO ENSAIO DE FASE 1B DO RYCALl® ARM210 PARA O TRATAMENTO DE MIOPATIAS RELACIONADAS AO RECEPTOR DE RIANODINA 1
ARMGO Pharma, Inc. (ARMGO), uma empresa privada no setor biofarmacêutico que desenvolve uma nova classe de drogas de moléculas pequenas ou micro moléculas conhecidas como Rycals®, anunciou em 29 de janeiro de 2024 a publicação dos resultados de um estudo de Fase 1b de seu Rycal ARM210 (também conhecido como S48168), para o tratamento de miopatias relacionadas ao receptor 1 de Ryanodina (RYR1-RM), uma doença muscular órfã, também chamada de “doença rara”, por ser uma doença que afeta uma pequena percentagem da população.
2024 a publicação dos resultados de um estudo de Fase 1b de seu Rycal ARM210 (também conhecido como S48168), para o tratamento de miopatias relacionadas ao receptor 1 de Ryanodina (RYR1-RM), uma doença muscular órfã, também chamada de “doença rara”, por ser uma doença que afeta uma pequena percentagem da população.
Os dados foram publicados em um artigo intitulado 'Rycal S48168 (ARM210) para Miopatias Relacionadas ao RYR1: um ensaio de fase um, estudo em aberto, e de ensaio de escalonamento de dose', de autoria do Dr. Joshua Todd et al, no Journal eClinicalMedicine, parte da família de publicações Lancet. O artigo revisa os dados do estudo de Fase 1b do ARM210 e seu novo mecanismo de ação alostérico (MoA) visando a causa raiz da doença relacionada ao RYR-1 (RYR1-RM): mutação do Receptor 1 de Ryanodina (RYR1).
O gene RYR1 que codifica o Receptor 1 de Ryanodina RYR1, um canal intracelular de liberação de cálcio, vaza em doenças musculares. Vazamentos intracelulares de cálcio causados por canais RYR1 com mutação prejudicam a contração muscular, levando à fraqueza muscular e perda de função, e ativam vias tóxicas que danificam os músculos, causando os sintomas das doenças relacionadas ao RYR1.
O ensaio de Fase 1b, aberto, de escalonamento de dose confirmou a segurança, tolerabilidade e farmacocinética da dosagem de 120 e 200 mg de ARM210 diariamente durante 29 dias em homens e mulheres adultos afetados pelas doenças relacionadas ao RYR1 (RYR1-RM).
É importante ressaltar que o estudo também demonstrou eficácia preliminar no grupo de dose mais alta em dois sintomas característicos das doenças relacionadas ao RYR1 (RYR1-RM): 1) alívio significativo da fadiga avaliada pelo sistema PROMIS-fatigue (Patient-Reported-Outcome Measurement Information System) t-scores, e 2) melhora da força dos proximais avaliada pelo exame físico de abdução do ombro (Medical Research Council Grading). Estes resultados justificam o desenvolvimento futuro do ARM210 como um potencial tratamento e modificador da doença para as miopatias relacionadas ao RYR1 (RYR1-RM) em um ensaio de Fase 2 randomizado e controlado por placebo.
O ensaio de Fase 1b concluído foi conduzido em colaboração com o National Institute of Neurological Disorders and Stroke (NINDS) e National Institute of Health (NIH) sob um Acordo Cooperativo de Pesquisa e Desenvolvimento (CRADA - Cooperative Research and Development Agreement ), com o apoio da Fundação RYR-1, Pittsburgh, PA, EUA.

(Foto esquerda para direita: Dr. Tokunbor Lawal (autor da publicação), Dr. Mike Goldberg (co-fundador/Co-presidente de Pesquisas da Fundação RYR-1), e Dr. Payam Mohassel (Pesquisador Principal e Autor Senior)).
“Estamos muito satisfeitos com os resultados do ensaio com as doenças relacionadas ao RYR1 (RYR1-RM) conduzido em conjunto com o NIH, pois o estudo confirmou a segurança e tolerabilidade do ARM210, mas o mais importante, demonstrou pela primeira vez que o nosso Rycal®, ARM210, pode reverter os sintomas desta doença muscular crônica e devastadora em um curto período de tratamento. Isso é muito promissor”, disse Gene Marcantonio, M.D., Ph.D., CEO da ARMGO Pharma. “Esperamos, portanto, continuar rapidamente no desenvolvimento do ARM210 para levar este primeiro e potencial tratamento aos pacientes com as doenças relacionadas ao RYR1, com o apoio da Fundação RYR-1 e da comunidade de pacientes.”
Michael F. Goldberg, MD, MPH, co-presidente de pesquisa da Fundação RYR-1 acrescentou: “Estamos entusiasmados com a publicação deste importante estudo, pois representa um farol de esperança para muitos indivíduos e famílias de todo o mundo afetados pelas doenças relacionadas ao RYR1. Estamos ansiosos pelas próximas etapas de ensaios no desenvolvimento dessa importante droga.”
Mais informações sobre este estudo de Fase 1b podem ser encontradas online em: https://clinicaltrials.gov/study/NCT04141670. O ensaio foi apoiado pelos Programas de Pesquisa Intramural do NIH/NINDS, NIH/NINR, um NIH Clinical Center Bench to Bedside Award (2017-551673) e pelo parceiro de colaboração anterior da ARMGO, Les Laboratoires Servier. O conteúdo é de responsabilidade exclusiva dos autores e não representa necessariamente a opinião oficial dos Institutos Nacionais de Saúde.
BREVE REVISÃO SOBRE A FUNÇÃO DO RYR1 E DOENÇAS RELACIONADAS
Os RYRs são canais homotetraméricos de liberação de cálcio intracelular responsáveis pelo fluxo de Ca2+ dos retículos sarcoplasmáticos/endoplasmáticos (RS/RE) para o citoplasma da maioria dos tipos de células. RYR1 é a isoforma predominante no músculo esquelético de mamíferos, onde a liberação de Ca2+ via RYR1 é necessária para o acoplamento excitação-contração e função muscular normal. Já o RYR2 é a isoforma predominante no músculo cardíaco onde a liberação de Ca2+ via RYR2 é necessária para a função normal do músculo cardíaco. Mutações genéticas humanas nos genes RYR1 e RYR2 fazem com que o cálcio vaze dos canais RYR, levando à doença (Figura 1).
Os canais RYR normalmente alternam entre um estado de repouso (fechado) e excitado (aberto). Em certas doenças, em que existe a mutação genética, o RYR é modificado e torna-se incontinente ou seja, fica vazando. No caso do RYR1, este desempenha um papel crítico no músculo esquelético, e as mutações do RYR1 em humanos levam a uma miopatia progressiva, conhecida como doenças relacionadas ao RYR1 (RYR1-RM), privando o músculo da capacidade de responder eficazmente aos sinais de contração, levando à fraqueza muscular.
Sobre o Rycals® - A inibição do canal interromperia o vazamento, mas esta intervenção geralmente não seria benéfica, uma vez que bloquearia a função normal do RYR. O Rycals®, são moléculas que podem restaurar a função normal do canal sem bloquear o RYR, e abrem a possibilidade de intervenções terapêuticas.
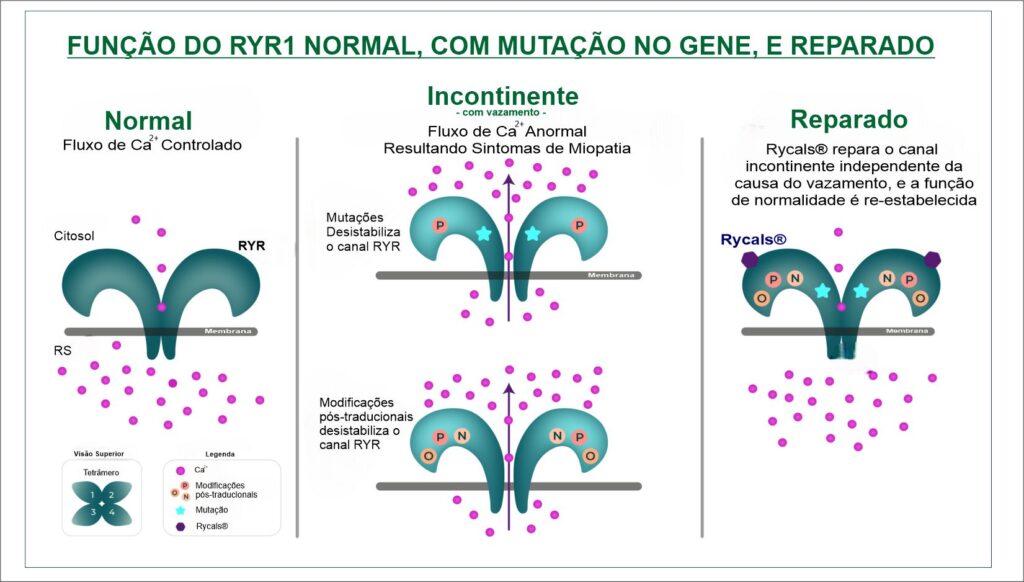
Figura 1: Modelo da função do RYR. O RYR controla o fluxo de cálcio (Ca2+) de dentro do retículo sarcoplasmático (RS) através da membrana até o citoplasma. (Quadro Esquerdo) O RYR normal regula o fluxo de cálcio alternando entre um estado fechado e aberto. (Quadro Meio) Na RYR1-RM, o RYR mutado apresenta vazamento, levando ao fluxo anormal de cálcio, resultando em sintomas da doença. Modificações pós-traducionais* (MPTs) do RYR agravam ainda mais o vazamento. Em outros distúrbios musculares, como DMD, insuficiência cardíaca ou sarcopenia, as modificações pós-traducionais (MPTs) do RYR podem causar vazamento no canal, levando a um fluxo anormal de cálcio, resultando em sintomas da doença. (Quadro Direito) Rycals® liga-se aos canais incontinentes (com vazamento) e repara o vazamento independentemente da causa do vazamento, restaurando a função normal do canal. O Rycals® não bloqueia o RYR.
*As modificações pós-traducionais (MPTs) são modificações químicas e estruturais de uma cadeia proteica após a sua tradução. Estas modificações podem determinar a atividade, a localização e interações com outras proteínas.
A Miopatia Congênita Centronuclear (MCCN) é uma doença muscular congênita rara caracterizada por fibras celulares com núcleos centralizados proeminentes em biópsias musculares. A doença é clinicamente heterogênea, variando de fenótipos hipotônicos graves já no nascimento até fraqueza muscular leve com início na idade adulta, e pode ter múltiplos modos de herança em associação de causa por mutações nos genes MTM1, DNM2, BIN1 e RYR1.
Assim como existe uma grande complexidade no diagnóstico de uma miopatia, tema esse abordado em outra postagem no SORRYR-1.com.br, essas diferentes causas, são também motivo de grande confusão em diagnósticos. É importante consultar um médico especialista para um diagnóstico preciso e assim obter informações detalhadas sobre a mutação específica no caso individual. As diferentes mutações genéticas que causam a Miopatia Congênita Centronuclear podem resultar em variações na gravidade, evolução e sintomas da doença. É importante dizer que a gravidade dos sintomas da doença pode variar de pessoa para pessoa, mesmo com a mesma mutação genética.
Seguem as diferentes causas de origem da Miopatia Congênita Centronuclear (MCCN):
O gene MTM1 é responsável pela codificação de proteína chamada miotubularina. Essa proteína desempenha um papel importante na função muscular, fundamental por atuar como uma enzima fosfatase de desempenho crítico na regulação do tráfego de vesículas dentro das células musculares, particularmente nas fibras musculares esqueléticas. Quando há uma mutação no gene MTM1, a produção ou função da miotubularina é afetada, e sso pode resultar em um acúmulo anormal de vesículas dentro das fibras musculares, levando à fraqueza muscular e outros sintomas associados à Miopatia Congênita Centronuclear.
A dinamina 2, codificada pelo gene DNM2, desempenha um papel crucial na regulação do tráfego de vesículas que transportam proteínas essenciais para a função muscular normal. Ela ajuda a controlar a fusão e divisão dessas vesículas, permitindo a entrega adequada de proteínas contráteis, como a miosina e actina, aos locais onde são necessárias para a contração muscular.
Quando ocorrem mutações no gene DNM2, a função da dinamina 2 pode ser comprometida, resultando em distúrbios que pode levar a fraqueza muscular, em especial nos músculos proximais, e outros sintomas associados a condições da MCCN. Portanto, a dinamina 2 desempenha um papel importante na manutenção da função muscular saudável.
O gene BIN1 codifica a proteína anexina A2, que está envolvida na regulação das membranas celulares e no tráfego de vesículas nas células musculares. Essas funções desempenhadas pela anexina A2 são essenciais para a saúde e a função das fibras musculares, a mutação nesse gene afeta negativamente a estrutura e a função das células musculares, o que resulta em fraqueza muscular e outros sintomas associados à CCNM. A gravidade e a apresentação dos sintomas podem variar com base na mutação específica do gene BIN1 envolvida.
O gene RYR1 codifica o receptor de rianodina 1, que é uma proteína essencial para a função das fibras musculares. O receptor de rianodina está envolvido na liberação (válvula de controle) de cálcio das reservas intracelulares, um processo fundamental para a contração e relaxamento muscular.
As doenças musculares são aquelas que afetam a estrutura e funcionamento do músculo, sendo as principais: as distrofias musculares, as miopatias congênitas, as miopatias inflamatórias e as miopatias endócrinas e metabólicas. É importante destacar que cada uma delas possui suas variações que também se diferenciam.
Essas doenças já foram muito confundidas em diagnósticos no passado, e fico triste, porque isso ainda tem acontecido nos dias de hoje, mesmo com os avanços científicos. A única razão que acredito ser ainda a causa para essa confusão nesses diagnósticos seria por essas doenças serem consideradas “doenças raras”, portanto, muitas vezes desconhecidas por parte da comunidade médica. Assim, pode haver a falha no momento dos exames clínicos, ponto inicial para diagnóstico de qualquer doença.
Eu mesmo vivi uma experiência dessa, pois no decorrer de grande parte da minha vida eu recebi vários “diagnósticos” de Distrofia Muscular Congênita (DMC) do tipo: Duchenne, Facioescapuloumeral e Cinturas. E, os prognósticos foram do pior a até o mais brando. Estes diagnósticos ou hipóteses de diagnósticos vieram até de importantes instituições, como de uma clínica indicada pelo MDA (Muscular Distrophy Association), maior referência ligada a essa doença.
Deve-se levar em consideração que, naquela época, pouco se sabia sobre essa doença, nem tão pouco sobre a genética humana; contudo, um erro de diagnóstico hoje em dia seria inaceitável. Essa situação causou em mim grandes transtornos, de emocional aos físicos. Somente aos 44 anos de idade foi que finalmente obtive meu correto e “definitivo diagnóstico”, ou seja, de que sou portador de Miopatia Congênita Centronuclear (MCC), causada pela mutação no gene RYR1.
A Distrofia Muscular Congênita e a Miopatia Congênita Centronuclear apresentam várias características em comum, tais como: são doenças de origem genética, afetam os músculos esqueléticos, caracterizam-se clinicamente por hipotonia e fraqueza muscular, geralmente apresentam-se desde o nascimento, têm curso clínico estático ou lentamente progressivo. Essas doenças não tem cura, e o tratamento envolve terapia de suporte, como fisioterapia, dispositivos de mobilidade e, em alguns casos, medicamentos. Mesmo assim, as duas doenças neuromusculares diferem entre si.
Daí, eu volto com a questão sobre as falhas nos diagnósticos, já que muitos médicos se prendem somente ao resultado do exame genético e não conhecerem os sinais clínicos das diferentes doenças e particularidades dos indivíduos afetados.
Assim, essas noções devem ser levadas em consideração por três razões: Primeiro, muitas das miopatias congênitas podem ser causadas por mutações em mais de um gene, o que sugere um impacto da heterogeneidade genética. Segundo, mutações no mesmo gene podem causar diferentes patologias musculares. Terceiro, a mesma mutação genética pode levar a diferentes características patológicas e sintomatológicas em membros da mesma família ou no mesmo indivíduo em idades diferentes.
Em resumo, eu destacaria que tanto a Distrofia Muscular, quanto a Miopatia Congênita Centronuclear são de origem genética, mas distintas em termos de suas características clínicas e podem variar em gravidade de pessoa para pessoa. Enquanto a Distrofia Muscular envolve a degeneração progressiva dos músculos devido a problemas na estrutura das proteínas musculares, a Miopatia Congênita Centronuclear é caracterizada por uma anormalidade na localização dos núcleos das células musculares. Assim, é importante consultar um médico especialista para um diagnóstico preciso, para que se possa ser feito um acompanhamento adequado do caso, pois o tratamento pode variar dependendo da condição clínica específica de cada indivíduo.

