A Miopatia Congênita Centronuclear é uma doença que causa muitas exclamações, mas também, interrogações. Sempre que as pessoas chegam até mim para questionar sobre minha doença, elas fazem uma exclamação dizendo, “nunca tinha ouvido sobre sua doença !”, e em seguida questionam como ela me afeta, e mais uma vez elas exclamam dizendo, "puxa vida !", e por ultimo exclamam novamente, “essa é uma super rara !”, daí respondo, "sim, no pé da palavra, ela é uma Doença Rara ou talvez uma Doença Ultrarrara.
A Miopatia Congênita Centronuclear decorrente de mutações no gene RYR1 é classificada como Doenças Raras ou Doenças Ultrarraras ?
Entender essas condições é essencial para oferecer o suporte adequado e assegurar abordagens terapêuticas personalizadas tanto para o paciente quanto para seus familiares. Doenças raras nem sempre recebem a atenção que merecem porque afetam relativamente poucas pessoas. Muitas vezes, pode levar anos para que uma pessoa receba o diagnóstico correto de uma doença rara, e cerca de 95% das doenças raras e ultrarraras ainda não têm tratamento.
Diferentes partes do mundo, as doenças raras e ultrarraras são identificadas e tratadas de maneiras diferentes, mas de um modo genérico a definição é mais ou menos assim:
Uma Doença Rara é aquela que acomete no máximo 65 pessoas a cada 100.000 habitantes, o que corresponde a uma prevalência aproximada de 1 para cada 1.500 indivíduos. A maior parte dessas condições tem origem genética, embora algumas possam surgir devido a fatores infecciosos ou relacionados ao sistema imunológico.
A Doença Ultrarrara, como o próprio nome indica, corresponde a condições ainda mais incomuns, com uma incidência de cerca de 1 caso para cada 50.000 indivíduos. Por serem extremamente infrequentes e pouco conhecidas, essas doenças geralmente apresentam maior dificuldade diagnóstica e exigem cuidados altamente especializados e personalizados.
Com relação à questão levantada no início do texto, a prevalência exata da Miopatia Congênita Centronuclear causada especificamente pela mutação no gene RYR-1 é desconhecida.
A dificuldade em determinar um número exato ocorre porque:
• Ela é uma doença ultrarrara por estar dentro de um grupo de doenças já raras, as Miopatias Congênitas.
• Os dados epidemiológicos costumam ser apresentados para a Miopatia Congênita Centronuclear (MCCN) considerando o conjunto das patologias que compõem esse grupo, que inclui mutações nos genes MTM1, DNM2, BIN1 e RYR1.
• O gene RYR1 está associado a um espectro de doenças musculares conhecidas como Doenças Relacionadas ao RYR1 ou RYR1-DR, sendo a forma Centronuclear apenas uma das possíveis manifestações, juntamente com a Miopatia Central Core (MCC), e outras.
Embora não haja um número específico de prevalência para a Miopatia Congênita Centronuclear causada pela mutação no RYR1, podemos contextualizar com base nas informações disponíveis para as condições relacionadas:
• No grupo da Miopatia Congênita Centronuclear (MCCN) em geral, a prevalência geral do grupo é desconhecida. A forma mais comum dentro deste grupo é a Miopatia Miotubular (ligada ao X) causada por mutações no gene MTM1, cuja incidência é estimada em cerca de 1 em 50.000 indivíduos do sexo masculino.
• No grupo das Doenças Relacionadas ao RYR1 (RYR1-DR) em geral, a doença mais frequentemente associado é a Miopatia Central Core (MCC), que é a forma mais comum de miopatia congênita não distrófica.
Um estudo de uma série pediátrica na Espanha estimou a incidência de Miopatias Relacionadas ao RYR1, que abrange a Centronuclear e a Central Core, se mostra em cerca de 1 em 10.000 nascidos vivos na área de estudo.
Em síntese, a mutação no gene RYR1 está associada a um fenótipo de ocorrência rara. A prevalência específica do subtipo Centronuclear (MCCN) ainda não foi estabelecida de forma isolada na literatura médica, sendo considerada extremamente baixa. Entretanto, não há consenso atual sobre classificá-la como no grupo de Doenças Raras ou no grupo de Doenças Ultrarraras.
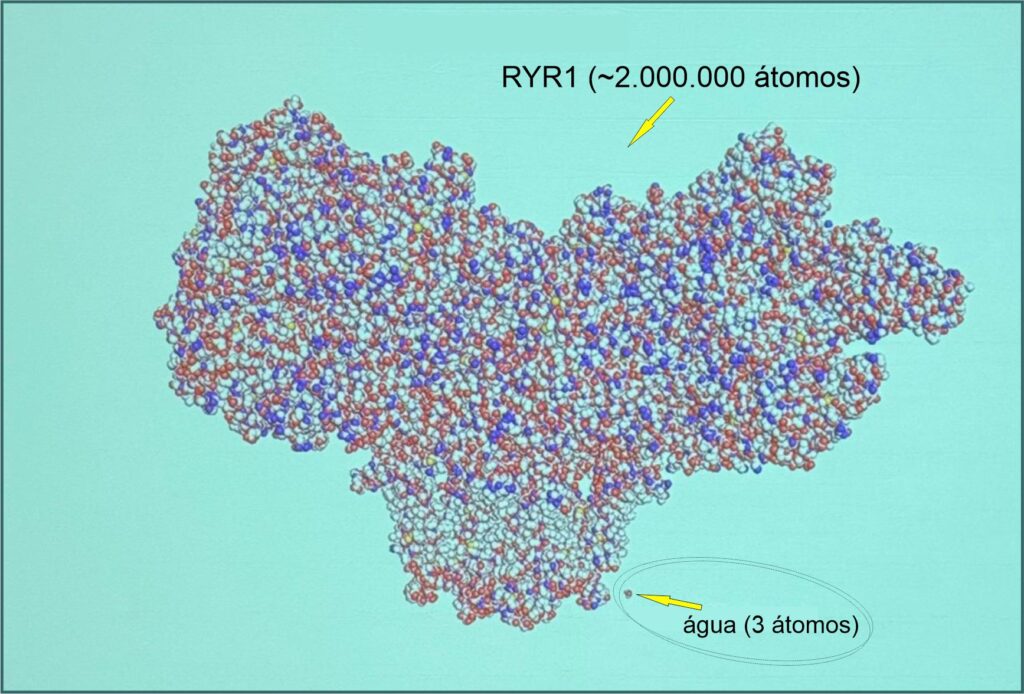
O RYR1 é um dos maiores genes do corpo humano, e é responsável para o funcionamento dos nossos músculos.
O gene RYR1 (Receptor de Rianodina 1) é um dos maiores e mais complexos genes do genoma humano. Ele contém mais de 100 exons e cerca de 15 mil pares de bases na sua sequencia de DNA, abrangendo uma grande extensão no cromossomo 19. A estimativa é que o gene RYR1 tenha cerca de 2 milhões de átomos, e esse é somente um cálculo de aproximação, pois a quantidade exata pode variar dependendo da sequência específica de nucleotídeos.
Para se ter uma idéia da complexidade do RYR1, ele é imensamente maior e mais complexo do que moléculas simples como da água, tão crucial para nossa vida, e a título de comparação, pasmem ! …o RYR1 tem 2 milhões de átomos, e a água, também conhecida como H20, tem somente 3 átomos.
O gene RYR1 que é crucial para a função do receptor de rianodina, que é um canal de cálcio localizado no Retículo  Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+
Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+) do RS para o citoplasma; 4). O cálcio dispara a contração muscular ao permitir a interação Actina-Miosina; 5). Para o relaxamento, o sinal cessa, o RYR1 se fecha, e rapidamente retorna o cálcio do citoplasma, devolvendo-o ao RS.
Em resumo, o RYR1 atua como o principal portão de saída do cálcio do reservatório intracelular. Ele é o mediador essencial que garante que o comando elétrico do nervo seja traduzido em movimento mecânico, controlando a entrada abrupta de cálcio que inicia a contração e, consequentemente, permitindo que a sua reabsorção finalize o processo para o relaxamento.
Como disse no início deste texto, devido ao tamanho e complexidade do RYR1, mutações nesse gene podem estar associadas a várias doenças musculares, incluindo:
O estudo científico do RYR1 é importante para entender essas condições ou doenças, e desenvolver tratamentos adequados em busca de minimizar seus efeitos e sintomas, pensando até na sua própria cura. Além disso, a análise genética pode ajudar em diagnósticos precoces e na prevenção de complicações associadas a essas doenças.
Durante o tempo que tenho acompanhado a movimentação de informações em torno das questões relacionadas ao RYR1, observo um grande esforço da comunidade científica em busca de entender o mecanismo de funcionamento deste complexo e grandioso gene, grande no tamanho, mas sobretudo grande em sua importância. Essas questões fazem com que o desafio ainda seja maior não só por conta dos indivíduos que têm uma doença a ele relacionada, mas também para compreensão sobre o impacto do envelhecimento celular em qualquer outro indivíduo. Observo também um grande movimento de trabalhos sendo desenvolvidos por cientistas de toda parte do mundo em busca de desenvolver uma droga para tratamentos adequados, para de repente ao menos minimizar seus efeitos e sintomas, até sua cura das doenças relacionadas ao RYR1. Particularmente, minha percepção é que as pesquisas através da técnica de reposicionamento de drogas, também chamado de redirecionamento de fármacos têm se mostrado mais promissoras no curto prazo. O “reposicionamento de drogas” se trata de uma estratégia que busca descobrir novas aplicações terapêuticas para medicamentos já existentes, que originalmente foram desenvolvidos para tratar outras doenças. Essa técnica tem a vantagem por seus reduzidos custos, assim como pela redução de tempo gasto no processo de desenvolvimento, e menores riscos no uso da eventual droga, pois já se conhece a farmacocinética e toxicidade do medicamento pesquisado, um exemplo real e prático de pesquisa em curso está se dando com o Sulfato de Salbutamol.
Contudo, a terapia genética também tem se mostrado uma abordagem promissora para o tratamento e cura de diversas doenças genéticas, oferecendo a possibilidade de corrigir ou substituir genes defeituosos. Embora este tipo de tratamento ainda esteja em desenvolvimento e não seja uma solução universal para algumas doenças, como por exemplo certas formas de miopatias ou doenças hereditárias, os resultados das pesquisas têm sido encorajadores. No entanto, a eficácia e a segurança da terapia genética podem variar dependendo da doença, do tipo de terapia utilizada, e condições do paciente. Além disso, questões éticas e de custo também são consideradas importantes. Contudo, enquanto a terapia genética oferece uma expectativa otimista, é uma área em evolução que requer mais pesquisas, altos recursos financeiros, e testes clínicos para se consolidar como a melhor solução para todas as doenças genéticas.
No caso específico das doenças relacionadas à mutação do gene RYR1, a terapia genética apresenta vários desafios, principalmente, como escrito no início deste texto, devido ao tamanho do gene, complexidade das doenças relacionadas a ele, mas também por considerar que os músculos representam aproximadamente 40% da massa total do corpo humano, e a maior parte dessa massa muscular é composta por músculos esqueléticos. Observe a seguir algumas das dificuldades:
Esses desafios tornam a pesquisa e o desenvolvimento de terapias para mutações no gene RYR1 complexos e exigem abordagens inovadoras e multidisciplinares.
Eu, enquanto portador de uma doença causada pela mutação nesse “grandioso” RYR1, tenho uma relação muito particular com esse gene, e a cada dia que passa o conheço um pouco mais, ..... confira minha próxima postagem intitulada “Eu vi o meu gene em 3D e entendi o que acontece dentro de mim”.
Buscando estar sempre conectado com as notícias relacionadas e de interesse dos portadores de mutação no gene RYR1, mais especificamente a Miopatia Congênita Centronuclear, que se trata da doença que me acomete, tomei conhecimento da recente pré-publicação científica, intitulada, “O Propofol liga-se diretamente e inibe o receptor 1 de rianodina do músculo esquelético (RYR1)”, versão postada em 12 de janeiro de 2024, na qual os pesquisadores, Thomas T. Joseph, MD, PhD¹, Weiming Bu, PhD¹, Omid Haji-Ghassemi, PhD², Yu Seby Chen, PhD², Kellie Woll, PhD², Paul D. Allen, MD, PhD⁴, Grace Brannigan, PhD³, Filip Van Petegem, PhD², Roderic G. Eckenhoff, MD¹, através de resultados obtidos em estudos e ensaios sugerem em seus relatos que o propofol, um agente anestésico intravenoso de curta ação, em concentrações clínicas, se liga ao receptor de rianodina tipo 1 (RYR1), inibindo sua abertura, podendo assim, prevenir as manifestações clínicas da Hipertermia Maligna (HM), mesmo com exposição a agentes desencadeantes como os anestésicos voláteis.
Confira a integra da publicação pelo seguinte link: https://www.biorxiv.org/content/10.1101/2024.01.10.575040v1.full
O receptor de rianodina tipo 1 (RYR1) desempenha um papel central na determinação de quando (tempo), e quanta (quantidade) força é produzida pelos músculos esqueléticos, que é necessária e essencial para movimentação do corpo e atividades físicas diárias dos indivíduos. Como principal canal de liberação de íon de cálcio (Ca²⁺) no retículo sarcoplasmático do músculo esquelético, a mutação genética no receptor de rianodina tipo 1 (RYR1), tem a ela subjacentes, algumas doenças ou distúrbios musculares, tais como a Miopatia Centronuclear, Miopatia Central Core, Miopatia Multi-Minicore, Desproporção Congênita do Tipo de Fibra, incluindo distúrbios como a Rabdomiólise por Esforço, e a Hipertermia Maligna (HM).
Em pacientes com a mutação no gene RYR1, “a crise” de Hipertermia Maligna, causada pela exposição a algumas drogas desencadeantes, como os anestésicos voláteis halogenados, pode direcionar o RYR1 a deixar o canal de rianodina em um estado aberto, resultando em uma liberação descontrolada de Ca²⁺, acarretando em tensão no sarcômero, e consequente produção de calor. A restauração de Ca²⁺ no retículo sarcoplasmático também consome ATP (adenosina trifosfato), molécula responsável pelo depósito de energia celular, gerando também por consequência uma carga metabólica adicional insustentável.
Ao anestesiar pacientes com mutações genéticas conhecidas pela suscetibilidade a Hipertermia Maligna, o anestésico geral intravenoso não desencadeante propofol é comumente substituído por anestésicos desencadeantes. As evidências de ligação direta de agentes anestésicos ao RYR1 ou seus parceiros de ligação são escassas, e as interações em nível atômico do propofol com o RYR1 são totalmente desconhecidas. Os pesquisadores mostram no trabalho acima descrito que o propofol diminui a abertura do receptor do canal de rianodina (RYR1) com vesículas no retículo sarcoplasmático e bicamadas lipídicas planas, e que inibe a liberação de Ca²⁺ induzida por ativador do retículo sarcoplasmático no músculo esquelético humano. Além de confirmar a ligação direta, a marcação por fotoafinidade usando m-azipropofol (AziPm) revelou vários supostos locais de ligação de propofol no RYR1. A projeção pela simulação dinâmica da afinidade de ligação molecular sugere que o propofol se liga a pelo menos um destes locais em concentrações clínicas. Esses achados convidam à hipótese de que, além de o propofol não desencadear a Hipertermia Maligna, ele também pode ser protetor contra a Hipertermia Maligna, inibindo o fluxo induzido de Ca²⁺ através do canal de rianodina - RYR1.
¹ Department of Anesthesiology and Critical Care, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA USA; ² Department of Biochemistry, University of British Columbia, Vancouver, BC, Canada; ³ Department of Physics and Center for Computational and Integrative Biology, Rutgers University, Camden, NJ USA; ⁴ Department of Anesthesiology, University of Tennesee, Knoxville, TN USA
O artigo acima mencionado foi publicado na bioRxiv, repositório aberto de pré-publicação direcionado as ciências biológicas (https://www.biorxiv.org/), e hospedado pelo Cold Spring Harbor Laboratory (CSHL).
As doenças relacionadas ao RYR1 são identificadas com base em sua classificação histopatológica, isto é, pela aparência da biópsia do músculo na lâmina do microscópio. A diferenciação encontrada na biópsia designará o tipo da doença, se é por exemplo, Miopatia Central Core, Miopatia Multiminicore, Miopatia Centronuclear, ou Desproporção Congênita de Tipos de Fibras.
A Miopatia Congênita Centronuclear (MCCN) e a Miopatia Central Core (MCC) são ambas doenças musculares hereditárias causadas por mutações genéticas. A MCCN pode estar associada a mutações em diferentes genes, no DNM2, BIN1, MTM1, e RYR1, já a MCC está associada somente ao gene RYR1.
Esses tipos de doenças relacionadas ao RYR1 variam amplamente em termos dos seus diferentes sinais e sintomas, de quando eles inicialmente se apresentaram, além da sua respectiva gravidade. Embora sejam altamente variáveis, os sintomas presentes também dependem se a mutação do gene RYR1 é autossômica dominante ou autossômica recessiva.
Uma pergunta que sempre chega até mim é sobre as diferenças entre a Miopatia Centronuclear e a Miopatia Central Core. Assim, eu, enquanto portador da Miopatia Congênita Centronuclear (MCN), buscarei esclarecer pontualmente neste texto, me atendo ao tipo que me acomete, que é a pela mutação no gene RYR1.
Estas doenças apesar de terem sintomas parecidos e compartilharem de algumas características clínicas em comum, se confundem entre si, e apresentam com algumas diferenças distintas:
Miopatia Congênita Centronuclear (MCCN)
Miopatia Central Core (MCC)
Em resumo, a Miopatia Congênita Centronuclear e a Miopatia Central Core, ambas doenças relacionada ao RYR1, têm seus sintomas e sinais físicos que podem se parecer, podem se confundir, mas são diferentes, a contar da análise histológica das células musculares em uma biópsia, exame este que é crucial para diferenciar entre as duas condições e determinar o diagnóstico correto, conduta médica, tratamento, e até prognóstico de evolução.
Nota: todo o material escrito nesta página é oriundo de pesquisa científica, e conclusões próprias do autor deste website, que convive como portador, desde o nascimento, com a Miopatia Congênita Centronuclear causada pela mutação no gene RYR-1
A Miopatia Congênita Centronuclear (MCCN) é uma doença muscular congênita rara caracterizada por fibras celulares com núcleos centralizados proeminentes em biópsias musculares. A doença é clinicamente heterogênea, variando de fenótipos hipotônicos graves já no nascimento até fraqueza muscular leve com início na idade adulta, e pode ter múltiplos modos de herança em associação de causa por mutações nos genes MTM1, DNM2, BIN1 e RYR1.
Assim como existe uma grande complexidade no diagnóstico de uma miopatia, tema esse abordado em outra postagem no SORRYR-1.com.br, essas diferentes causas, são também motivo de grande confusão em diagnósticos. É importante consultar um médico especialista para um diagnóstico preciso e assim obter informações detalhadas sobre a mutação específica no caso individual. As diferentes mutações genéticas que causam a Miopatia Congênita Centronuclear podem resultar em variações na gravidade, evolução e sintomas da doença. É importante dizer que a gravidade dos sintomas da doença pode variar de pessoa para pessoa, mesmo com a mesma mutação genética.
Seguem as diferentes causas de origem da Miopatia Congênita Centronuclear (MCCN):
O gene MTM1 é responsável pela codificação de proteína chamada miotubularina. Essa proteína desempenha um papel importante na função muscular, fundamental por atuar como uma enzima fosfatase de desempenho crítico na regulação do tráfego de vesículas dentro das células musculares, particularmente nas fibras musculares esqueléticas. Quando há uma mutação no gene MTM1, a produção ou função da miotubularina é afetada, e sso pode resultar em um acúmulo anormal de vesículas dentro das fibras musculares, levando à fraqueza muscular e outros sintomas associados à Miopatia Congênita Centronuclear.
A dinamina 2, codificada pelo gene DNM2, desempenha um papel crucial na regulação do tráfego de vesículas que transportam proteínas essenciais para a função muscular normal. Ela ajuda a controlar a fusão e divisão dessas vesículas, permitindo a entrega adequada de proteínas contráteis, como a miosina e actina, aos locais onde são necessárias para a contração muscular.
Quando ocorrem mutações no gene DNM2, a função da dinamina 2 pode ser comprometida, resultando em distúrbios que pode levar a fraqueza muscular, em especial nos músculos proximais, e outros sintomas associados a condições da MCCN. Portanto, a dinamina 2 desempenha um papel importante na manutenção da função muscular saudável.
O gene BIN1 codifica a proteína anexina A2, que está envolvida na regulação das membranas celulares e no tráfego de vesículas nas células musculares. Essas funções desempenhadas pela anexina A2 são essenciais para a saúde e a função das fibras musculares, a mutação nesse gene afeta negativamente a estrutura e a função das células musculares, o que resulta em fraqueza muscular e outros sintomas associados à CCNM. A gravidade e a apresentação dos sintomas podem variar com base na mutação específica do gene BIN1 envolvida.
O gene RYR1 codifica o receptor de rianodina 1, que é uma proteína essencial para a função das fibras musculares. O receptor de rianodina está envolvido na liberação (válvula de controle) de cálcio das reservas intracelulares, um processo fundamental para a contração e relaxamento muscular.
As doenças musculares são aquelas que afetam a estrutura e funcionamento do músculo, sendo as principais: as distrofias musculares, as miopatias congênitas, as miopatias inflamatórias e as miopatias endócrinas e metabólicas. É importante destacar que cada uma delas possui suas variações que também se diferenciam.
Essas doenças já foram muito confundidas em diagnósticos no passado, e fico triste, porque isso ainda tem acontecido nos dias de hoje, mesmo com os avanços científicos. A única razão que acredito ser ainda a causa para essa confusão nesses diagnósticos seria por essas doenças serem consideradas “doenças raras”, portanto, muitas vezes desconhecidas por parte da comunidade médica. Assim, pode haver a falha no momento dos exames clínicos, ponto inicial para diagnóstico de qualquer doença.
Eu mesmo vivi uma experiência dessa, pois no decorrer de grande parte da minha vida eu recebi vários “diagnósticos” de Distrofia Muscular Congênita (DMC) do tipo: Duchenne, Facioescapuloumeral e Cinturas. E, os prognósticos foram do pior a até o mais brando. Estes diagnósticos ou hipóteses de diagnósticos vieram até de importantes instituições, como de uma clínica indicada pelo MDA (Muscular Distrophy Association), maior referência ligada a essa doença.
Deve-se levar em consideração que, naquela época, pouco se sabia sobre essa doença, nem tão pouco sobre a genética humana; contudo, um erro de diagnóstico hoje em dia seria inaceitável. Essa situação causou em mim grandes transtornos, de emocional aos físicos. Somente aos 44 anos de idade foi que finalmente obtive meu correto e “definitivo diagnóstico”, ou seja, de que sou portador de Miopatia Congênita Centronuclear (MCC), causada pela mutação no gene RYR1.
A Distrofia Muscular Congênita e a Miopatia Congênita Centronuclear apresentam várias características em comum, tais como: são doenças de origem genética, afetam os músculos esqueléticos, caracterizam-se clinicamente por hipotonia e fraqueza muscular, geralmente apresentam-se desde o nascimento, têm curso clínico estático ou lentamente progressivo. Essas doenças não tem cura, e o tratamento envolve terapia de suporte, como fisioterapia, dispositivos de mobilidade e, em alguns casos, medicamentos. Mesmo assim, as duas doenças neuromusculares diferem entre si.
Daí, eu volto com a questão sobre as falhas nos diagnósticos, já que muitos médicos se prendem somente ao resultado do exame genético e não conhecerem os sinais clínicos das diferentes doenças e particularidades dos indivíduos afetados.
Assim, essas noções devem ser levadas em consideração por três razões: Primeiro, muitas das miopatias congênitas podem ser causadas por mutações em mais de um gene, o que sugere um impacto da heterogeneidade genética. Segundo, mutações no mesmo gene podem causar diferentes patologias musculares. Terceiro, a mesma mutação genética pode levar a diferentes características patológicas e sintomatológicas em membros da mesma família ou no mesmo indivíduo em idades diferentes.
Em resumo, eu destacaria que tanto a Distrofia Muscular, quanto a Miopatia Congênita Centronuclear são de origem genética, mas distintas em termos de suas características clínicas e podem variar em gravidade de pessoa para pessoa. Enquanto a Distrofia Muscular envolve a degeneração progressiva dos músculos devido a problemas na estrutura das proteínas musculares, a Miopatia Congênita Centronuclear é caracterizada por uma anormalidade na localização dos núcleos das células musculares. Assim, é importante consultar um médico especialista para um diagnóstico preciso, para que se possa ser feito um acompanhamento adequado do caso, pois o tratamento pode variar dependendo da condição clínica específica de cada indivíduo.
Abordagem Respiratória na Miopatia Centronuclear
Por: Alessandra C. Dorça
As Miopatias Congênitas Centronucleares (CNMs) são um grupo de doenças neuromusculares hereditárias classificadas como miopatias congênitas. Embora vários genes causadores tenham sido identificados, alguns pacientes não abrigam nenhuma das mutações atualmente conhecidas. Esses diversos distúrbios têm características histológicas comuns, que incluem uma alta proporção de fibras musculares com núcleo central, e atributos clínicos de fraqueza muscular e insuficiência respiratória.
Neste texto será abordado especificamente sobre questões respiratórias. As alterações respiratórias podem se manifestar inicialmente durante o sono, mas sintomas diurnos e dificuldade de proteção da via aérea predominam à medida que a disfunção muscular respiratória evolui para mais grave.
A avaliação respiratória do indivíduo afetado é fundamental para acompanhar o comprometimento muscular. A capacidade muscular respiratória pode ser avaliada usando uma variedade de testes clínicos, como análise de capacidades pulmonares, forças musculares inspiratórias e expiratórias, e pico de fluxo de tosse.
O comprometimento respiratório costuma ser a maior causa de morte, já que algumas formas mais graves apresentam hipotonia e fraqueza de região faríngea, ocasionando distúrbios de deglutição e comprometimento respiratório recorrente devido a pneumonias bronco aspirativa. Nestes casos a indicação precoce da sonda gástrica pode ser de fundamental importância.
A disfunção dos músculos respiratórios pode ser agravada por restrições mecânicas que aumentam a carga respiratória, incluindo baixa complacência pulmonar, defeitos restritivos musculoesqueléticos na parede torácica e cifoescoliose. Com a evolução da doença, a fraqueza da musculatura bulbar ocasiona episódios recorrentes de engasgos, e a troca espontânea da consistência alimentar pode facilitar a mastigação favorece a deglutição. As manifestações funcionais da insuficiência dos músculos respiratórios podem incluir distúrbios do sono, fadiga, tosse ineficaz e hipoventilação.
As várias mutações genéticas das miopatias possuem progressão diferente, mas a diminuição da capacidade motora ocasiona, por consequência a diminuição do volume corrente e do tempo inspiratório com aumento concomitante da carga elástica e diminuição comprimento-tensão musculares em volumes pulmonares mais altos. O resultado funcional é um padrão respiratório rápido e superficial, necessidade de suporte ventilatório para manter a capacidade elástica muscular, e evitar a restrição severa da caixa torácica.
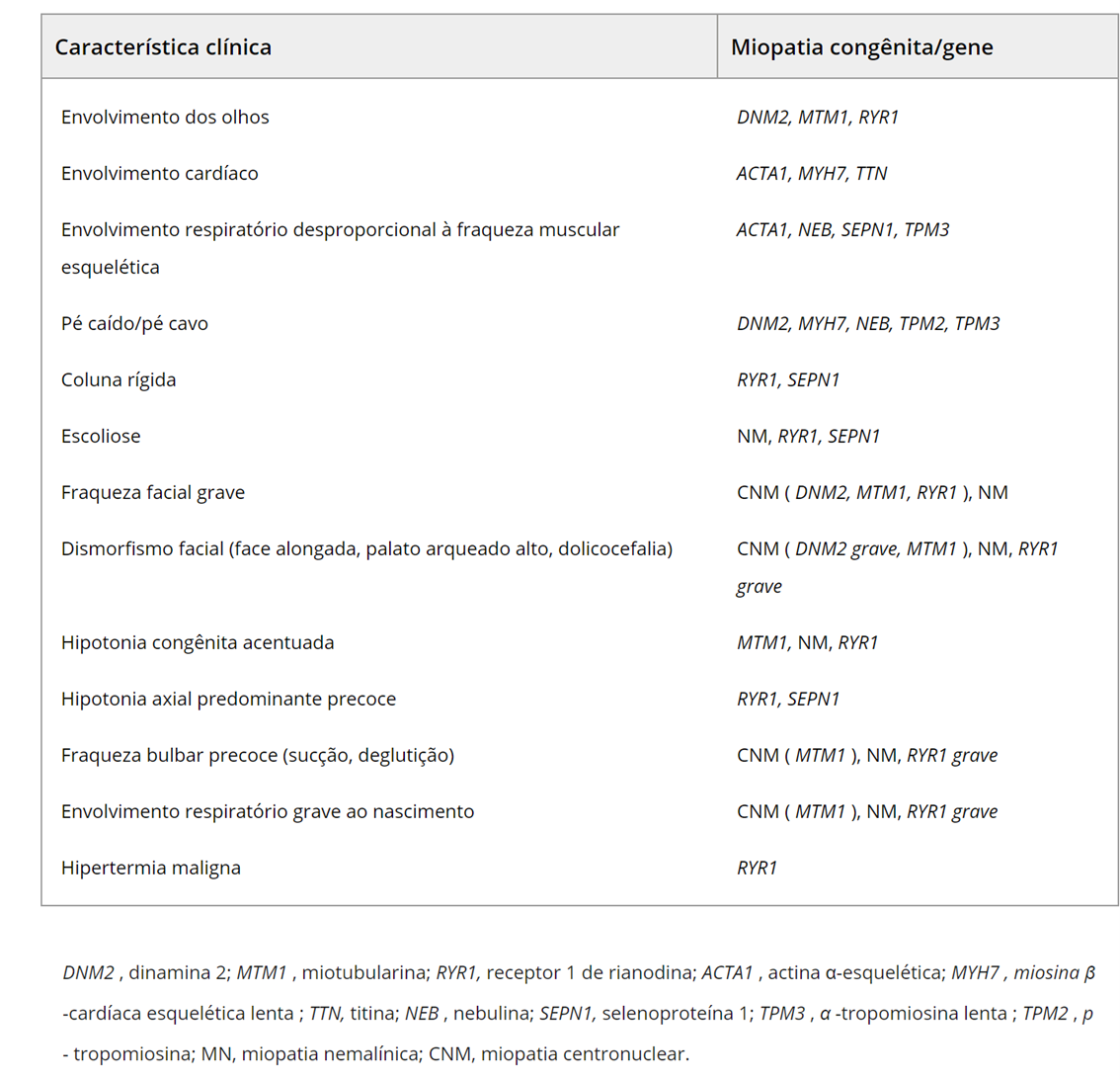
O quadro a seguir apresenta as características clínicas nas miopatias causadas por mutações nos diversos genes:
Cuidados Respiratórios nas Miopatias Congênitas Centronuclear (CNMs)
As várias mutações genéticas que causam as miopatias centronucleares apresentam progressões variadas, principalmente relativo à velocidade da evolução da doença. No caso, a progressão lenta ou mais acelerada traz a necessidade de cuidado respiratório próximo, fazendo assim necessário uma Avaliação Respiratória Funcional, visando uma objetiva e eficaz Abordagem Terapéutica Respiratória (tratamento).
Avaliação Respiratória Funcional
A avaliação respiratória funcional é capaz de acompanhar a velocidade de progressão da doença, e assim direcionar as abordagens necessárias em cada momento da doença, e deve ser realizada a cada 6 meses, desta forma as abordagens preventivas poderão minimizar as complicações da patologia, como segue:
1 - Avaliação da capacidade pulmonar
CVF (Capacidade Vital Forçada) - A prova de função pulmonar é capaz de predizer a capacidade vital e correlacionar com a idade e o peso, desta forma é possível verificar o comprometimento no volume pulmonar devido a diminuição da força dos músculos respiratórios. A CVF (Capacidade Vital Forçada) orienta a necessidade de suporte respiratório.
2 - As forças musculares respiratórias, PIMAX (pressão inspiratória máxima), e PEMAX (pressão expiratória máxima), são mensuradas através da Manovacuometria.
PIMAX - é a pressão inspiratória máxima, e reflete o valor da força muscular inspiratória, ou seja, a força diafragmática. O diafragma é o maior musculo da respiração e corresponde a 50 % da capacidade de geração de volume pulmonar. A fraqueza diafragmática favorece a hipoventilação noturna, a dificuldade de geração de volume pulmonar e restrição respiratória.
PEMAX - é a pressão expiratória máxima, e reflete o valor da força da musculatura abdominal, que é responsável pela centralização da força do tronco, auxilia a tosse e favorece o movimento de expiração e auxilia explosão da tosse, o que proporciona a melhor proteção pulmonar
3 - Pico de Fluxo de Tosse (PFT)
A tosse é o principal mecanismo de proteção da via aérea. Para a efetividade da tosse é necessário a sinergia de vários músculos da região laríngea, glote, e músculos abdominais. A avaliação do pico de fluxo de tosse é capaz de predizer a força desta musculatura, e a sincronia no movimento. O comprometimento da musculatura bulbar tem como resposta inicial a diminuição do pico de tosse.
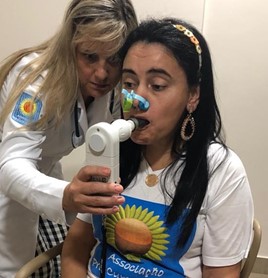
Avaliação respiratória das forças musculares ( Micro RPM), prova de função pulmonar (Mini Spir)
Abordagem Terapáutica Respiratória - Tratamento
A fisioterapia respiratória é responsável pela minimização dos comprometimentos respiratórios nas miopatias centronucleares, pois interfere na progressão da perda muscular, minimiza as complicações da perda de capacidade pulmonar, mantem a funcionalidade respiratória como fala, deglutição e tosse. Fazem parte das estratégias utilizadas no tratamento:
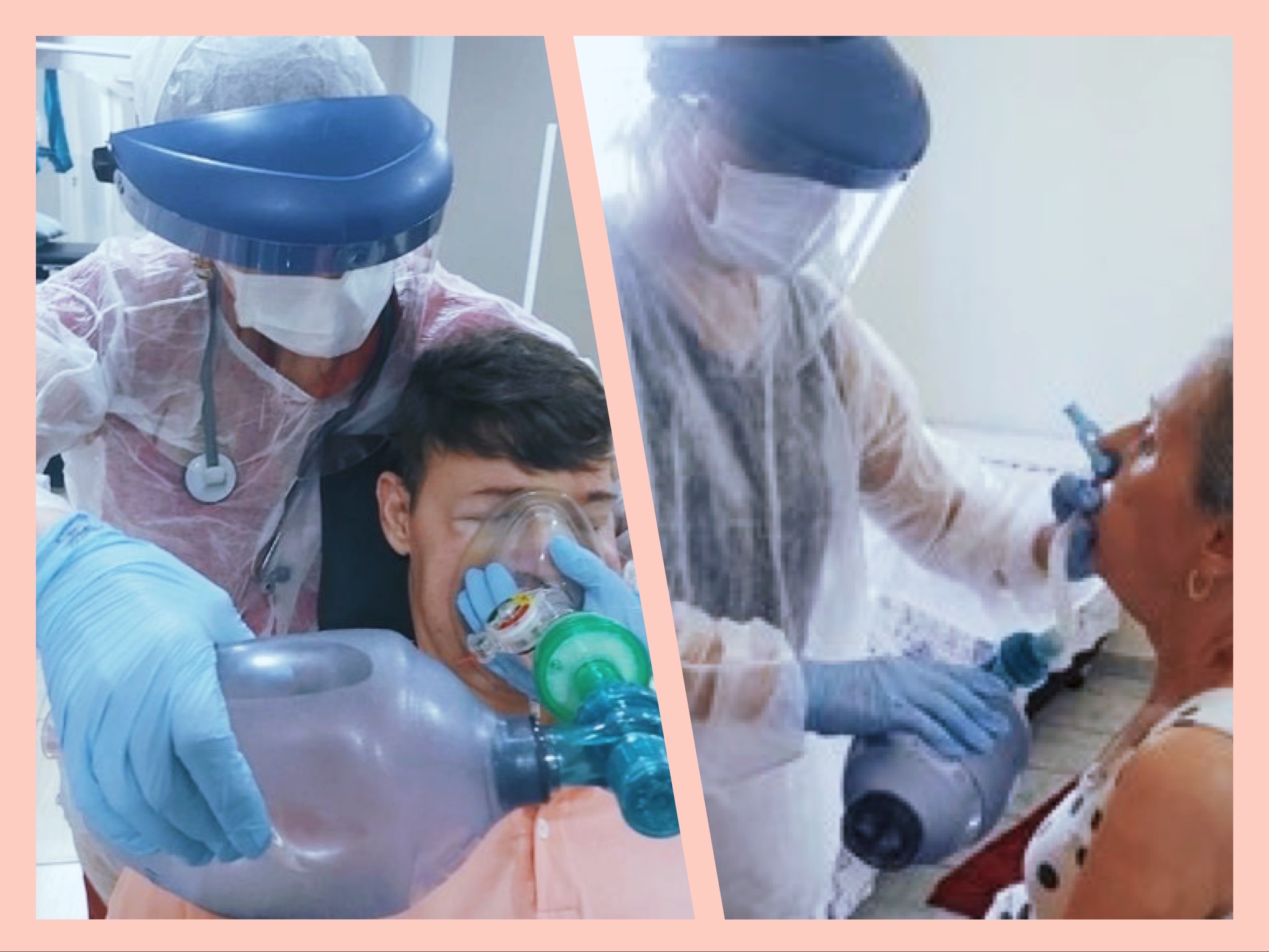
Fig 1 -Exercício de empilhamento de ar \ Fig 2 - Exercícios respiratórios para manutenção da capacidade pulmonar e treinamento da musculatura bulbar (ambos utilizando a bolsa de insuflação (ambu)
O cuidado respiratório deve ser uma prioridade nos portadores de Miopatia Centronucelar, e o profissional deve ser capaz de identificar a alteração funcional e correlacionar com a melhor terapia a ser aplicada, sendo esta a melhor estratégia para evitar maiores complicações para o paciente.
Bibliografia Consultada:
Smith BK, Goddard M, Childers MK. Avaliação respiratória nas miopatias centronucleares. Nervo Muscular 2014;50(3):315-326. doi:10.1002/mus.24249
Benditt JO. Respiratory Care of Patients With Neuromuscular Disease. Respir Care. 2019 Jun;64(6):679-688. doi: 10.4187/respcare.06827. PMID: 31110036.

