Hans Christian, um escritor dinamarquês conhecido mundialmente por suas histórias infantis, certa vez disse, "Onde as palavras falham, a música fala", e eu completo dizendo que a música é um meio de transporte capaz de dirigir nossas emoções e sensações a todos os sentimentos. Contudo, através da música pude também experimentar outros benefícios além do que normalmente esperado, e que vou contar a seguir qual foi…
Quando Luciano (meu filho) se foi aos 23 anos de idade, eu busquei de alguma forma me manter próximo dele alimentando as lembranças sobre ele. Luciano, parece ter vivido pouco tempo de vida, sim, mas ele fez muito… ele foi triatleta, músico multi-instrumentista, dentre outros feitos. Com esta vontade que tive de buscar estar perto dele, pensei, no esporte não vou conseguir viver o que ele viveu, daí decidi ir de encontro com ele pelo caminho da música e então tocar violino, seu principal instrumento musical desde os 6 anos de idade. Música sempre foi algo que fez parte do dia a dia de nossa família, e cremos que Deus além de usar a música para falar conosco, ele nos concedeu o dom da musicalidade, daí não vi maiores dificuldades e resolvi embarcar nessa jornada. O Professor Ricardo Seoud embarcou comigo nesta grande empreitada para me ajudar a conquistar o objetivo de aprender a tocar violino, contudo eu sabia que antes iria precisar romper com minhas barreiras de dificuldade física para conseguir segurar o instrumento. Sendo assim, além do meu professor Seoud, contei com o trabalho conjunto de Lorena, minha fisioterapeuta, a qual me acompanhou fazendo as devidas orientações de técnicas de exercícios de consciência corporal, educação postural, alongamentos, e respiração.
Existe um fato interessante que liga minha relação como portador de deficiência física ao de tocar um instrumento musical. Quando era criança minha mãe me colocou na aula de violão, e mais tarde me deu uma flauta, não entendia o porque, mas e hoje pude concluir que mesmo com sua simplicidade sobre o entendimento da doença que me acometia, o que ela realmente buscava era promover a minha educação física, e com a flauta o meu condicionamento respiratório. Não existia conhecimento técnico que tocar um instrumento musical poderia funcionar como uma forma de fisioterapia. Naquela época, as minhas limitações físicas ainda não estavam tão severas como atualmente, hoje o simples fato de conseguir segurar o violino é o meu grande desafio. Lembro que nas primeiras aulas, Sissi, minha esposa, precisava colocar duas a três almofadas sob meu braço para servir de apoio de sustentação para conseguir segurar o instrumento, mas que no decorrer dos treinamentos eu superei e consegui manter o instrumento na posição correta, ou melhor, meio correta, e penso que foi Luciano quem estaria me ajudando fisicamente, não com o triathlon, mas com o simples ato de conseguir fisicamente segurar o violino.
Sabe-se através do conhecimento fisiológico, que o ato de tocar um instrumento musical envolve uma série de movimentos físicos que podem ajudar a melhorar a coordenação motora, a força muscular e a flexibilidade, além de estimular áreas do cérebro relacionadas à motricidade e à cognição. No mais, a música pode ter um efeito terapêutico, promovendo relaxamento, reduzindo o estresse e melhorando o bem-estar emocional.
É um entendimento, que toda forma de atividade física é crucial e benéfica para o condicionamento físico dos portadores de miopatia, inclusive como sendo a única e comprovada forma de tratamento. E no caso, pela minha experiência pessoal pude concluir que o ato de tocar um instrumento musical, é um exercício físico que atuou verdadeiramente como um complemento à minha rotina diária de fisioterapia proporcionando os seguintes benefícios:
Por fim, gostaria de ilustrar e celebrar esta postagem com uma música muito significativa para mim (senti A música escolhida, Londonderry Air, é uma ária publicada em 1855, e que se tornou como um hino irlandes, sendo considerada uma das canções mais tocadas no mundo, e que teve muitas versões de letras, mas a mais conhecida e autêntica foi Danny Boy, composta por um inglês em 1910. Sua melodia emotiva e letra tocante, fala sobre a despedida, e espera de um amor eterno, com uma mensagem de esperança e a promessa de que o amor por alguém persistirá além da vida.
...ao abrir o video no yoytube, click no canto direito inferior no icone ⌈ ⌉ para abrir a imagem em toda a tela
Responder perguntas relacionadas à miopatia que me acomete é um dos meus propósitos com o SorRYR-1. Uma pergunta que sempre me fazem, e a resposta sempre causa uma certa confusão a quem respondo é o fato de meu CPK ser normal. Acredito que isso se dá pela desinformação de muitas pessoas entender que as miopatias estão diretamente ligadas a uma alteração dos níveis de CPK.
O CPK, também conhecido como creatinoquinase, é uma enzima encontrada no tecido muscular. Os níveis elevados de CPK podem indicar danos ou lesões nos músculos, coração ou cérebro. No entanto, é importante considerar que os níveis de CPK podem variar de pessoa para pessoa, e existem também várias condições de saúde e atividades físicas que podem levar a um aumento temporário nos níveis de CPK. Uma outra questão que os cientistas ainda pesquisam, e não tem uma resposta, é sobre porque os níveis de CPK se comportam de forma diferente nas miopatias, em alguns individuos em níveis alterado, em outros normal.
Os níveis elevados de CPK podem ser indicativos de várias condições de saúde, como exemplo:
Em uma miopatia, os níveis de creatinoquinase (CPK ou CK) podem variar, dependendo do tipo e da causa da miopatia. Em alguns casos, os níveis de CPK podem estar elevados, enquanto em outros casos podem ser normais ou até mesmo baixos. Portanto, não há uma resposta definitiva sobre o porquê dos níveis de CPK estarem altos ou baixos em uma miopatia, assim, é importante consultar um médico para obter um diagnóstico preciso e interpretar corretamente os resultados dos exames.
No meu caso com diagnóstico de Miopatia Congênita Centronuclear, sempre tive meus resultados de exames sempre com o CPK no nível da normalidade, portanto, é importante ressaltar que o diagnóstico da miopatia não é baseado exclusivamente nos níveis de CPK, mas sim por uma combinação de sintomas clínicos, exames de imagens, exame genético, biópsia muscular, dentre outros exames.
Assim sendo, é importante lembrar que os níveis de CPK devem ser interpretados em conjunto com outros sintomas e exames clínicos. E caso haja suspeita de Miopatia, é fundamental realizar uma avaliação médica completa com um especialista em doenças neuromusculares para obter um diagnóstico preciso. O médico irá considerar todos os aspectos clínicos e resultados de exames para chegar a um diagnóstico correto.
As doenças musculares são aquelas que afetam a estrutura e funcionamento do músculo, sendo as principais: as distrofias musculares, as miopatias congênitas, as miopatias inflamatórias e as miopatias endócrinas e metabólicas. É importante destacar que cada uma delas possui suas variações que também se diferenciam.
Essas doenças já foram muito confundidas em diagnósticos no passado, e fico triste, porque isso ainda tem acontecido nos dias de hoje, mesmo com os avanços científicos. A única razão que acredito ser ainda a causa para essa confusão nesses diagnósticos seria por essas doenças serem consideradas “doenças raras”, portanto, muitas vezes desconhecidas por parte da comunidade médica. Assim, pode haver a falha no momento dos exames clínicos, ponto inicial para diagnóstico de qualquer doença.
Eu mesmo vivi uma experiência dessa, pois no decorrer de grande parte da minha vida eu recebi vários “diagnósticos” de Distrofia Muscular Congênita (DMC) do tipo: Duchenne, Facioescapuloumeral e Cinturas. E, os prognósticos foram do pior a até o mais brando. Estes diagnósticos ou hipóteses de diagnósticos vieram até de importantes instituições, como de uma clínica indicada pelo MDA (Muscular Distrophy Association), maior referência ligada a essa doença.
Deve-se levar em consideração que, naquela época, pouco se sabia sobre essa doença, nem tão pouco sobre a genética humana; contudo, um erro de diagnóstico hoje em dia seria inaceitável. Essa situação causou em mim grandes transtornos, de emocional aos físicos. Somente aos 44 anos de idade foi que finalmente obtive meu correto e “definitivo diagnóstico”, ou seja, de que sou portador de Miopatia Congênita Centronuclear (MCC), causada pela mutação no gene RYR1.
A Distrofia Muscular Congênita e a Miopatia Congênita Centronuclear apresentam várias características em comum, tais como: são doenças de origem genética, afetam os músculos esqueléticos, caracterizam-se clinicamente por hipotonia e fraqueza muscular, geralmente apresentam-se desde o nascimento, têm curso clínico estático ou lentamente progressivo. Essas doenças não tem cura, e o tratamento envolve terapia de suporte, como fisioterapia, dispositivos de mobilidade e, em alguns casos, medicamentos. Mesmo assim, as duas doenças neuromusculares diferem entre si.
Daí, eu volto com a questão sobre as falhas nos diagnósticos, já que muitos médicos se prendem somente ao resultado do exame genético e não conhecerem os sinais clínicos das diferentes doenças e particularidades dos indivíduos afetados.
Assim, essas noções devem ser levadas em consideração por três razões: Primeiro, muitas das miopatias congênitas podem ser causadas por mutações em mais de um gene, o que sugere um impacto da heterogeneidade genética. Segundo, mutações no mesmo gene podem causar diferentes patologias musculares. Terceiro, a mesma mutação genética pode levar a diferentes características patológicas e sintomatológicas em membros da mesma família ou no mesmo indivíduo em idades diferentes.
Em resumo, eu destacaria que tanto a Distrofia Muscular, quanto a Miopatia Congênita Centronuclear são de origem genética, mas distintas em termos de suas características clínicas e podem variar em gravidade de pessoa para pessoa. Enquanto a Distrofia Muscular envolve a degeneração progressiva dos músculos devido a problemas na estrutura das proteínas musculares, a Miopatia Congênita Centronuclear é caracterizada por uma anormalidade na localização dos núcleos das células musculares. Assim, é importante consultar um médico especialista para um diagnóstico preciso, para que se possa ser feito um acompanhamento adequado do caso, pois o tratamento pode variar dependendo da condição clínica específica de cada indivíduo.
O diagnóstico de uma doença rara impõe ao indivíduo afetado além das consequências físicas a ela inerentes, alterações na sua rotina de vida. Esse mesmo diagnóstico pode pôr fim a um período de vida marcado por incertezas e ansiedades, muitas vezes em razão de uma demorada e sofrida peregrinação por consultas e exames médicos. Entretanto, com o diagnóstico em mãos começa-se uma nova etapa, que é a busca por respostas acerca da doença em questão, e que muitas vezes o médico não conseguiu responder. E o maior desafio em meio a essas buscas se dá por um tratamento eficiente visando a cura, ou ao menos de um que seja pelo alívio do seu sofrimento físico. Portanto, para o paciente essa dura realidade do diagnóstico, é normalmente dividida em duas etapas, uma que acontece no consultório médico, e a outra etapa, que se vive dentro das "quatro paredes da sua intimidade". O processo de assimilação do diagnóstico pode ser assustador e cheio de interrogações, e o indivíduo afetado tende buscar por si só as respostas que mais anseiam, tais como sobre prognóstico, cura, e tratamento. Essas buscas e pesquisas são normalmente feitas na internet, contudo, muitas vezes ao contrário de esclarecer a situação, pode torná-la mais confusa.
Entendo perfeitamente a reação desses indivíduos afetados, assim como de seus familiares, uma vez que o desconhecimento por muitos médicos sobre essas doenças ainda é bem limitado. Enfim, na prática, uma vez diante do diagnóstico que você é portador de uma doença que nunca havia ouvido falar a respeito anteriormente, e que ouve do médico, pessoa esta que se imaginava ser a quem poderia lhe ajudar, lhe dizer que a tal doença é rara, que se tem muito pouca informação sobre ela, e que para a mesma não tem cura e nem mesmo tratamento eficaz, e que seu prognóstico é de progressividade, …daí você como parte interessada, o que lhe resta ? … além das buscas na internet por informações, o maior desejo é encontrar outras pessoas que estejam vivendo com o mesmo diagnóstico. O SORRYR-1 tem se prestado a este propósito, tanto que tenho sido procurado por indivíduos afetados e/ou por seus familiares questionando sobre o que tenho feito para lidar com a doença nestes meus 60 anos de vida. Nas primeiras vezes em que fui questionado, confesso que tive dificuldade em responder, primeiro porque entendo que cada pessoa é única, e o que funciona para uma pessoa pode não funcionar para outra, mas com o tempo pude entender que o meu posicionamento pessoal poderia ser algo complementar ao que me propus com o SORRYR-1. Assim sendo, em atendimento a algumas sugestões, decidi escrever de maneira prática sobre o que entendo ser o mais importante para conviver com as doenças relacionadas ao RYR1, e especificamente no meu caso a Miopatia Congênita Centronuclear.
1 - Inicialmente falo sobre minha convicção pessoal dizendo que creio viver esta situação segundo um propósito, me baseando na palavra do apóstolo Paulo em Romanos 12:2 que diz, “.... transformem-se pela renovação da sua mente, para que sejam capazes de experimentar e comprovar a vontade de Deus para sua vida.” , entendendo que apesar das imposições a mim colocada pela doença, busco reinventar e viver um estilo de vida segundo minhas possibilidades, com espírito de resiliência e superação, e busco servir ao próximo como sendo este o propósito de Deus para minha vida.
2 - Atitudes positivas devem ser a palavra de ordem na sua maneira de viver. Assim, entender que o bem-estar emocional é fundamental para uma vida saudável e feliz, faz com que não deixe que sua condição física interfira no seu estado emocional. É importante também lembrar que todos enfrentamos desafios e dificuldades em diferentes aspectos de nossas vidas, e a deficiência física é apenas uma parte de quem somos. Portanto diante do espírito de resiliência e auto-estima positiva, busque se superar nas suas possibilidades e habilidades, não se limitando nas suas limitações, ao contrário, as potencializando.
3 - A atividade física é primordial, é nosso único tratamento disponível, seja fisioterapia ou ginástica, e essa prática deve ser parte de nossa rotina diária. Vale lembrar que nossas células musculares, pela mutação que temos no RYR1 não funciona da maneira correta (contração e relaxamento), interferindo assim em questões relacionadas ao nosso fortalecimento e movimentação, além de poder causar contraturas, rigidez, dores, fadiga, dentre outras. A atividade física, mesmo que passiva, pode nos ajudar com a qualidade de vida e até impedir a progressão da doença. Lançando mão da frase dita por meu neurologista, Dr Acary, que diz, “o RYR1 é movimento”, portanto, movimente, mexa-se, nunca pare de mexer, mexa do músculo do dedo do pé até os da face, assim, mexa-se sempre. Contudo, vale ressaltar que devemos estar atentos ao nosso limite, evitando o excesso de esforço físico e cansaço, pois isso pode agravar os sintomas da miopatia;
4 - O condicionamento respiratório também deve ser um grande ponto de atenção diante das dificuldades musculares generalizadas que enfrentamos. O enfraquecimento da musculatura do tórax, traqueia e diafragma pode interferir em questões sistêmicas. Assim, destaco como exemplo a dificuldade na expectoração de eventuais secreções, deglutição, e adequada troca de gases feita pelos pulmões. Leia mais sobre este tema na postagem “Abordagem Respiratória na Miopatia Centronuclear”;
5 - Outro ponto que deve ser observado é com nosso ganho de peso, uma vez que o quê se ganha nesa situação é a gordura, e como nosso tecido muscular que já não é lá essas coisas, ele é substituído com facilidade pelo tecido gorduroso. O aumento de peso dificulta ainda mais nossa capacidade em fazer atividade física, que por sua vez impede a queima de calorias, favorecendo assim ainda mais o acúmulo de gordura em nosso corpo. Esse acúmulo de gordura em nosso corpo, além de afetar nossa qualidade de vida, pode ainda nos trazer consequências patológicas graves, tais como cardiopatias, diabetes, hipertensão arterial, doenças no fígado, alguns tipos de câncer, problemas renais, dentre outros.
6 - Considerando nossa reduzida capacidade em fazer atividade física, temos uma propensão à fragilidade óssea, portanto, temos que evitar as quedas pelo alto risco de fraturas. Na prática, é que para uma fratura óssea, o tratamento deverá ser desde a imobilização do membro afetado, até a intervenção cirúrgica (observar riscos cirúrgicos), e em ambas situações ficaremos impedidos de movimentação física, o que causará ainda mais perda muscular.
7 - Evite contrair qualquer doença, seja um simples resfriado, ou qualquer outra doença. Por exemplo, nosso corpo diante de uma enfermidade com causa viral ou bacteriana, reage com uma resposta imunológica, liberando uma série de proteínas, as citocinas, as quais produzem uma reação inflamatória não apenas no local da infecção, mas também em outros órgãos, incluindo músculos e articulações, que são nosso maior ponto de atenção e fragilidade. Assim, com esse exemplo procurei mostrar porque uma simples gripe pode nos causar ainda mais fraqueza.
8 - Mantenha um estilo de vida praticando hábitos saudáveis, tais como, seguir uma dieta mais nutritiva, equilibrada, e saudável, ingerindo bastante água (35 ml/kg de peso corporal por dia), procure dormir o suficiente e com qualidade, se exponha periodicamente ao sol (8 às 11 horas), enfim, evite o estresse e preocupação excessiva, assim como hábitos nocivos a saúde física (cigarro, droga, e álcool).
9 - Fique atento e evite tratamentos experimentais, e medicamentos que não os indicados por seu médico especialista. Por exemplo, alguns medicamentos, como estatinas, anti-inflamatórios não esteroides (AINEs), e outros mais, podem piorar a miopatia.
10 - Faça regularmente um acompanhamento médico com especialista visando monitorar não somente sua miopatia, mas também sua condição física geral. É muito importante que os portadores de miopatia conversem com seu médico para discutir os fatores que podem piorar sua condição, e que possam aprender a gerenciá-los.
A FUNDAÇÃO RYR-1 REALIZOU EM JULHO DE 2022, EM PITTSBUGH, EUA, O PRIMEIRO WORKSHOP INTERNACIONAL DE PESQUISAS SOBRE DOENÇAS RELACIONADAS AO RYR-1
A Fundação RYR-1 realizou nos dias 21 e 22 de julho último, em Pittsburgh, nos Estados Unidos, o primeiro Workshop Internacional de Pesquisas sobre Doenças Relacionadas ao RYR-1, para discutir desde os mecanismos ao tratamento da doença. O evento reuniu 45 pesquisadores presenciais e 10 virtuais de 11 países diferentes, os quais se encontram desenvolvendo trabalhos em busca de tratamento ou cura da doença, e que na oportunidade puderam compartilhar entre eles suas respectivas pesquisas.
A organização do encontro convidou também 10 indivíduos afetados pelas doenças relacionadas ao RYR1, estando eu entre eles, para relatar nossas diferentes histórias de vida em relação à doença. O objetivo foi compartilhar nossa vivência, conhecimentos e interagir com os presentes, visando assim colaborar no desenvolvimento de novas estratégias para encontrar terapias, assim como também sensibilizar os pesquisadores sobre nosso anseio urgente por alguma forma de resultado prático de suas pesquisas para amenizar sintomas e até curar a doença.
No meu testemunho de vida diante da doença, em que relatei acontecimentos desde meu nascimento até os dias de hoje, teve três pontos que percebi chamar a atenção dos presentes. Um dos pontos foi sobre a exaustão sofrida em função do longo período de 44 anos que se levou para o diagnóstico. Essa mesma demora de tempo para diagnostico não seria uma realidade nos dias de hoje, mas ainda deve ser considerada como alta, pois sabe-se que atualmente uma “doença rara” leva em média de 7 a 10 anos para ser diagnosticada. No meu caso, a Miopatia Congênita Centronuclear não é uma das doenças mais comuns dentro do espectro das relacionadas a mutação do RYR-1, dificultando assim o seu diagnóstico. Outro ponto importante destacado foi dizer que mesmo recebendo o diagnóstico da doença, a falta de informação dado ao seu pouco conhecimento médico-científico, me causou o que disse ter sido uma espécie de cura emocional, gerando um grande estímulo positivo em busca de respostas ao desconhecido da doença. Entendo que a falta de diagnóstico e informações sobre uma doença, muitas vezes é pior do que seus próprios sintomas, e que o emocional tem um grande poder sobre o físico. E por fim, disse sobre o grande  momento vivido depois de 10 anos do diagnóstico, em que conheci a Fundação RYR-1, momento aquele em que ainda buscava informação sobre a doença. Essa organização tem o objetivo promover a troca de experiências entre os participantes, além de apoiar cientistas que trabalham no desenvolvimento de medicamentos e tratamento, a qual mostrou a mim que a Miopatia Congênita Centronuclear não era um campo tão desconhecido como pensava, e me fez acreditar em algo que eu nunca tinha pensado antes, que era a existência próxima de um medicamento para tratar ou aliviar os efeitos das Doenças relacionadas ao RYR1.
momento vivido depois de 10 anos do diagnóstico, em que conheci a Fundação RYR-1, momento aquele em que ainda buscava informação sobre a doença. Essa organização tem o objetivo promover a troca de experiências entre os participantes, além de apoiar cientistas que trabalham no desenvolvimento de medicamentos e tratamento, a qual mostrou a mim que a Miopatia Congênita Centronuclear não era um campo tão desconhecido como pensava, e me fez acreditar em algo que eu nunca tinha pensado antes, que era a existência próxima de um medicamento para tratar ou aliviar os efeitos das Doenças relacionadas ao RYR1.
Encontros como as conferências de famílias, e esse primeiro workshop de pesquisadores, me fazem entender que apesar de raro ou em meio a poucos, não me encontro sozinho, e assim além de aprender com os outros sobre como conviver com a doença, alimenta a expectativa em saber que existem médicos e cientistas em adiantado estágio de pesquisa por uma droga para tratamento e até a sua cura.
UM TESTEMUNHO DE VIDA DE UM PORTADOR DE MIOPATIA CONGENITA CENTRONUCLEAR EM MEIO ÀS ADVERSIDADES DA DOENÇA
A Fundação RYR-1 sediará em julho de 2022 o primeiro Workshop Internacional de Pesquisa em Doenças Relacionadas ao RYR-1, reunindo um grupo internacional de especialistas, cientistas pesquisadores, assim como um seleto grupo de indivíduos afetados por uma doença relacionada ao RYR-1. Neste encontro deverá acontecer um intercambio de informações sobre os mecanismos das doenças relacionadas ao RYR1, a posição atual sobre as pesquisas em curso e perspectivas futuras de tratamentos, mas o mais importante para nós afetados pela doença, é que teremos a oportunidade de expor nossas experiências, queixas, como a doença evolui e nos afeta no dia a dia.
Eu, como portador de Miopatia Congênita Centronulear, fui um dos indivíduos convidados a participar deste encontro, e pretendo na oportunidade oferecer insights pessoais visando ajudar os pesquisadores e clínicos a entender melhor como essa doença afeta nossos corpos nas várias fases e situações da vida. Para melhor conhecimento dos cientistas e pesquisadores presentes, nós convidados, deveremos fazer uma apresentação pessoal, como um histórico de toda a vida sobre nossa relação com a doença.
Em última análise, a partir de nossas histórias, fico na esperança que os cientistas com a compreensão aprimorada, possam mediante as informações recebidas incrementar suas pesquisas para produção de drogas e terapias eficazes em nosso benefício.
Atendendo a sugestão de pessoas que previamente tomaram conhecimentos do meu histórico de vida, reproduzo abaixo o texto base sobre o que será minha exposição no referido evento, o tornando assim de conhecimento de todos.
Meu nome é Orlando, brasileiro, moro em Goiânia, GO, cidade localizada na região centro oeste do Brasil. Minha história de vida começa em 1963, e este texto está focado na minha saúde física. Gostaria de começar dizendo que apesar de não ter sofrido complicações durante a gravidez da minha mãe, nem problemas no parto, meu desenvolvimento físico foi marcado desde o início da minha vida pelo atraso motor e pela hipotonia já notada no primeiro ano de vida. Vale ressaltar que eu já tinha um casal de irmãos fisicamente normais, livres de doenças.
Na época, em meados da década de 1960, diante das limitações científicas e do pouco conhecimento médico disponível em Goiânia, cidade onde morava, aos 7 anos, meus pais decidiram ir em busca de explicações e diagnóstico sobre o que estava me afetando fisicamente, e assim fomos para São Paulo, um grande centro médico do Brasil. Eu já apresentava marcos motores preocupantes, como a manobra de Gowers, dificuldade para subir degraus, dentre outros. Uma biópsia muscular veio comprovar relatando sinais inusitados e inespecíficos no tecido (somente coloração de H&E e trinômio de Masson), mas o neurologista limitou-se a dizer que eu estava acometido por uma potencial doença neuromuscular, e não foi indicado nenhum tratamento a ser realizado.
Em 1970, nasceu minha irmã, o que na época causou à família um misto de alegria pelo seu nascimento, mas preocupação porque ela já dava sinais claros de que também era acometida pela mesma doença inespecífica que me acometia. Meus pais, preocupados com a situação decidiram levar eu e minha irmã, ainda bebê, para outro grande centro médico, desta vez para o Departamento de Estudos Neurológicos da Universidade Federal do Rio de Janeiro. Lá, mais exames foram realizados, incluindo um eletromiografia, e desta vez nos foi dada uma hipótese de diagnóstico clínico de um tipo de Distrofia Muscular. Na ocasião o médico conversou com meus pais sobre as características e prognóstico daquela doença, assim como recomendou cuidados especiais com exercícios físicos para evitar uma possível progressão da doença, mas nada foi indicado como tratamento.
Durante minha infância e adolescência, mesmo considerando minhas dificuldades e as claras diferenças em relação às outras crianças, fui muito ativo, e tentei fazer tudo, nadei, andei de bicicleta, enfim, brinquei muito. Fui criado e ensinado de uma forma que não focava no que eu não podia fazer, mas no que eu queria e era capaz de fazer dentro das minhas habilidades. Ao longo dos anos, experimentei um padrão de piora física lenta, mas constante, tais como a dificuldade em subir escadas, levantar de uma cadeira, andar, manter o equilíbrio e riscos de queda. Emocionalmente também sofri muito por vezes me sentir diferente dos meus colegas, mas ao mesmo tempo em que fui fortemente apoiado pelos amigos e familiares, me fazendo sentir normal, mesmo sendo uma criança fisicamente anormal.
As dificuldades que sempre enfrentei na vida, me fizeram desenvolver um instinto de superação e de busca pela Independência. Casei-me muito jovem, aos 19 anos, quando ainda cursava faculdade. Já casado, aos 21 anos, mudamos para os EUA fazer meu MBA. Naquela época pensavaos em ter filhos, mas diante da falta de conhecimento e certeza sobre a doença que me acometia, por termos ouvido falar do MDA - Associação de Distrofia Muscular, fomos lá em busca de respostas e aconselhamento. Fomos a uma clínica associada em Austin, Texas, cidade onde morávamos, e consultados pelo Dr Jerry Tindel, que após uma bateria de exames, recebi o diagnóstico de Distrofia Muscular, sugerindo ser do tipo FSH-Facioscapulohumeral. Na consulta me foi explicado sobre a gravidade da doença, seu prognóstico, a probabilidade de ter um filho também afetado pela doença, e concluir dizendo que em poucos meses eu estaria em cadeira de rodas, contudo, nenhum tratamento me foi prescrito.
De volta ao Brasil, a vida continuou, e nos anos que se seguiram, as décadas de 1980 e 1990, minhas dificuldades físicas aumentaram, era sinal da evolução da miopatia, eu não conseguia mais me levantar de uma cadeira, e precisava usar uma bengala para apoiar minha caminhada e equilíbrio, mas ao contrário do que o médico do MDA tinha dito, eu não precisava de usar cadeira de rodas. Neste período estive muito envolvido com meus projetos profissionais e familiares. Eu tive um casal de filhos, e ambos nunca mostraram sinais de serem afetados pela doença. Luciano, meu filho mais velho, apesar de ter falecido aos 23 anos vítima de leucemia, durante sua vida como prova de sua capacidade física se tornou atleta de triathlon, e conquistou a medalha de bronze aos 20 anos no Campeonato Mundial de Triatlo em Vancouver, Canadá, o que demonstrou não ser portador de nenhuma miopatia. Priscila, nossa filha caçula, hoje é médica e casada, e é fisicamente normal, mas carrega uma mutação no gene RYR-1.
No ano de 2000, incomodado com a progressão e piora física com aumento da limitações impostas pela doença, porém não tão grave como dizia em 1985 o médico do MDA, fui novamente em busca de respostas, e desta vez acreditando na evolução científica marcada pela mudança do século. Procurei pelo que existia de mais avançado na ciência, e fui consultado pela Dra Mayana Zatz, geneticista do Departamento de Genética Humana da USP - Universidade de São Paulo, em busca de um diagnóstico, contudo, mais uma vez me viram como portador de uma Distrofia Muscular, mas novamente sem um laudo conclusivo.
Inconformado com a imprecisão dos hipotéticos diagnósticos que recebi no decorrer da minha vida, finalmente, em 2007, aos 44 anos, conheci o Dr Acary Bulle Oliveira, neurologista e chefe do Setor de Doenças Neuromusculares da UNIFESP - Universidade Federal de São Paulo, e sendo consultado por ele, finalmente recebi um diagnóstico clínico e conclusivo, corroborado por biópsia muscular que evidenciou anormalidades da rede intermiofibrilar típicas de uma Miopatia Congênita Centronuclear, bem como resultado de exame genético e pesquisa com sequenciamento total do exoma mostrando alteração (mutação) no gene RYR1.
Dada a raridade da miopatia que me afeta, minha idade e situação física, e evolução da doença, em 2017 fui convidado a visitar o NIH - National Institutes of Health, Bethesda, Maryland, EUA, e consultado pelo Dr Carsten Bonnemann, MD., Senior Investigator and Chief of the Neuromuscular and Neurogenetic Disorder me coloquei a disposição para participar de eventual estudo de pesquisa. Esse momento foi muito impactante na minha vida, pois pela primeira vez percebi que haviam pessoas interessadas na minha doença, e que já estavam desenvolvendo pesquisas em busca de tratamento e até de cura. Na época, outro grande marco foi ter sido apresentado à Fundação RYR1, que é uma associação que visa reunir pessoas afetadas por doenças relacionadas ao RYR1 de todo o mundo, promover a troca de experiências entre os participantes (afetados e familiares), além de apoiar cientistas e laboratórios que trabalham no desenvolvimento de medicamentos para tratamento, a até a terapia genética curativa. Enfim, toda essa experiência me fez ver e prever algo que nunca tinha pensado, que é a cura, ou pelo menos a existência de um medicamento para tratar ou aliviar os efeitos da Miopatia Congênita Centronuclear.
(este texto foi escrito originalmente em inglês, e traduzido para o português para esta postagem)
DOENÇAS RELACIONADAS AO RYR-1 É UM TERMO “GUARDA-CHUVA”, OU SEJA, QUE ABRANGE ALGUMAS DIFERENTES DOENÇAS MUSCULARES
As doenças relacionadas ao RYR1 são raras, e classificadas como doenças órfãs , “órfãs”, se trata de um termo usado para identificar uma doença que afeta uma pequena percentagem da população. A verdadeira prevalência dessas doenças é difícil de calcular, pois muitos casos são mal diagnosticados ou não diagnosticados. Também há relatos de prevalência ligeiramente aumentada em certas populações étnicas e geográficas.
As doenças relacionadas ao RYR1 são devidas a uma mutação ou mutações no gene RYR-1. Na prática, podemos sintetizar o processo de mutação da seguinte maneira: o gene RYR-1 codifica o receptor RYR-1, o qual é um canal de cálcio no retículo sarcoplasmático do músculo esquelético; o fluxo de cálcio através do receptor RYR-1 é um componente crítico para a excitação-contração muscular. Uma mutação no gene RYR-1 pode alterar o número, estrutura e/ou função do receptor RYR-1, podendo assim desencadear uma ampla gama de sintomas, e consequências clínicas diferentes, que podemos chamar de doenças relacionadas ao RYR1.
Historicamente, indivíduos com RYR1-RD eram diagnosticados com base em características da biópsia muscular, como núcleos centrais, bastonetes e desproporção do tipo de fibra, embora essas características não sejam exclusivas da RYR1-RD e possam mudar ao longo do tempo. Com o surgimento de mais doenças ligadas a variantes do RYR1 — como a síndrome de King-Denborough, rabdomiólise induzida por exercício e miopatias de início na idade adulta, a sobreposição diagnóstica aumentou. Por isso que para abranger a crescente gama de condições ou doenças relacionadas ao RYR1, incluindo casos de início na idade adulta recém-identificados, foi sugerido o uso do termo “doenças relacionados ao RYR1 (RYR1-RD)” como uma nomenclatura unificada para esse complexo espectro de doenças.
“Doenças Relacionadas ao RYR1 – RYR1-RD” é um termo abrangente que engloba uma gama de subtipos relacionados ao RYR1 que afetam o sistema neuromuscular em humanos. O Orphanet, ( https://www.orpha.net/ ) banco de dados europeu de doenças raras, lista diversas condições relacionadas ao gene RYR1 (figura a seguir):

Alguns pesquisadores classificam as doenças relacionadas ao RYR1 em vários subtipos com base nos seguintes critérios:
Achados de biópsia muscular (histopatologia):
Sintomas (fenótipo clínico):
Interações fármaco-gene (farmacogenética):
Os sintomas das doenças relacionadas ao RYR1 geralmente estão presentes desde o nascimento (congênitas) ou aparecem na primeira infância e podem ser estáticos, dinâmicos ou uma combinação de ambos. Os sintomas estáticos (presentes o tempo todo) incluem fraqueza muscular, atraso motor, dificuldades para andar e subir escadas, escoliose, fraqueza muscular facial e fraqueza muscular ocular (oftalmoparesia). Os sintomas dinâmicos (que aparecem e desaparecem com base em certos gatilhos) incluem doenças relacionadas ao calor, degradação muscular induzida por exercício (rabdomiólise), dor muscular (mialgia), cãibras musculares e fadiga.
As variantes do RYR1 também são a principal causa de suscetibilidade à hipertermia maligna (SHM), representando >60% dos casos. A hipertermia maligna (HM) é uma reação potencialmente fatal que ocorre em indivíduos suscetíveis após exposição a anestésicos voláteis ou relaxantes musculares despolarizantes, os quais desencadeiam um rápido aumento da temperatura corporal (hipertermia) e degradação muscular (rabdomiólise). As reações de HM são tratadas com o medicamento dantroleno.
Os sintomas apresentados por indivíduos com doenças relacionadas ao gene RYR1 podem ser bastante variáveis; no entanto, o curso da doença geralmente não é progressivo ou é muito lentamente progressivo. A expectativa de vida é geralmente normal em indivíduos afetados e o desenvolvimento cognitivo não é afetado. Embora não haja cura ou tratamento aprovado para doenças relacionadas ao gene RYR1, estratégias de suporte, incluindo fisioterapia, podem ajudar a controlar as limitações funcionais e promover uma alta qualidade de vida.
Como dito anteriormente, as Doenças Relacionadas a RYR-1 (RYR-1-RD) engloba diferentes tipos de doenças relacionados ao RYR1, e é também demonstrado como um termo “guarda-chuva” se referindo às suas subdivisões.
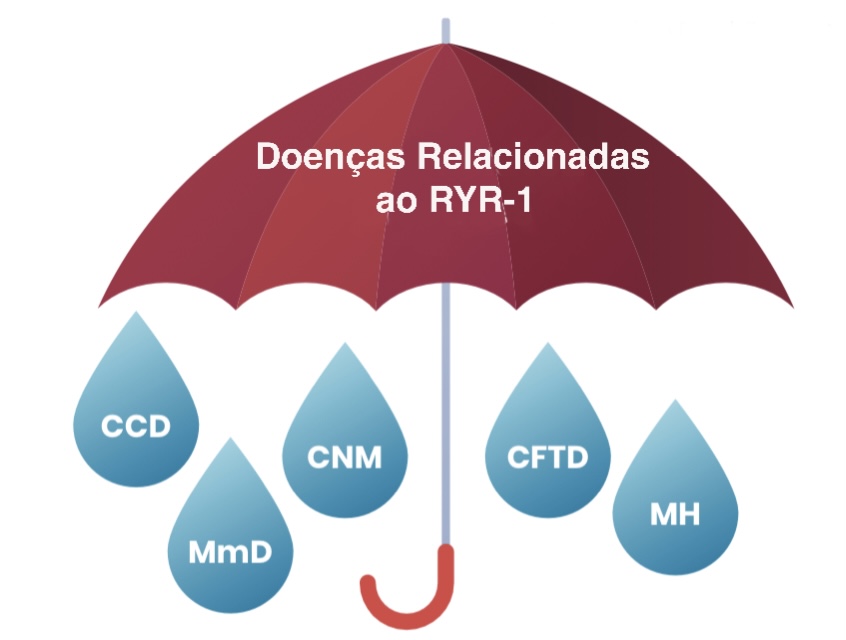
Miopatia Central Core - Central Core Disease (CCD)
Miopatia Central Core - Central Core Disease (CCD)
Descrita pela primeira vez por Magee e Shy em 1956. A DNC geralmente é herdada de forma dominante. Quando observadas ao microscópio, as fibras musculares com DNC apresentam coloração escura, mas também áreas claras no meio das fibras, que não são coradas. Essas áreas claras representam a ausência de atividade mitocondrial. As mitocôndrias são as estruturas responsáveis pela geração de energia para a célula.
A Miopatia Central Core (CCD) causa fraqueza muscular de leve a muito grave; no entanto, a maioria dos indivíduos afetados apresenta fraqueza muscular leve e persistente, que pode piorar com o tempo. Essa fraqueza afeta os músculos próximos ao tronco (músculos proximais), particularmente na parte superior das pernas e nos quadris. A fraqueza muscular também pode fazer com que os bebês afetados pareçam "flácidos" e resultar em atraso no desenvolvimento motor, como sentar, ficar em pé e andar. Bebês gravemente afetados apresentam tônus muscular profundamente fraco (hipotonia), resultando em dificuldades de alimentação e problemas respiratórios graves ou com risco de vida. A CCD também está associada a anormalidades esqueléticas, como curvatura excessiva da coluna vertebral (escoliose), luxação do quadril e deformidades articulares chamadas contraturas, que restringem o movimento de certas articulações. Indivíduos com CCD geralmente conseguem andar durante toda a vida.
Doença Multiminicore (MmD)
Doença Multiminicore (MmD) foi descrita pela primeira vez como doença multicore por Engel e colaboradores em 1971. A MmD é herdada de forma recessiva e causa fraqueza muscular e problemas de saúde relacionados, que variam de leves a potencialmente fatais. Quando observadas ao microscópio, as fibras musculares da MmD apresentam coloração escura, mas também várias áreas claras dentro de cada fibra muscular, sem coloração, resultando em uma aparência "roída por traças". Assim como na CCD, essas áreas sem coloração representam a ausência de atividade mitocondrial. Em geral, a MmD causa sintomas mais graves do que a CCD. Os pesquisadores identificaram quatro formas distintas de MmD:
Forma clássica: a forma mais comum, associada à fraqueza muscular no pescoço (axial) e tronco, com início na infância ou no início da infância, curvatura anormal da coluna vertebral (escoliose), comprometimento respiratório e hiperlaxidão (aumento da flexibilidade) das articulações dos membros.
Forma oftalmoplégica: associada à paralisia ou fraqueza dos músculos oculares, com fraqueza muscular generalizada e fraqueza facial grave.
Forma de início precoce: associada à artrogripose (contraturas articulares desde o nascimento).
Forma lentamente progressiva: associada ao comprometimento dos músculos das mãos.
A fraqueza muscular faz com que os bebês afetados pareçam "flácidos", com tônus muscular fraco (hipotonia), resultando em atraso no desenvolvimento motor, como sentar, ficar em pé e andar. A rigidez da parede torácica e da coluna vertebral também está associada à MmD. Quando combinada com a fraqueza dos músculos necessários para a respiração, podem ocorrer problemas respiratórios graves ou com risco de vida. Quase todas as crianças com MmD desenvolvem uma curvatura anormal da coluna vertebral (escoliose), que surge durante a infância e piora progressivamente com o tempo.
Miopatia Congênita Centronuclear - Centronuclear Myopathy (CNM)
A Miopatia Congênita Centronuclear (CNM) foi descrita pela primeira vez por Spiro e colaboradores em 1966, a MCN é herdada de forma recessiva. Ao serem examinadas ao microscópio, as fibras musculares da CNM apresentam núcleos (estruturas que contêm cromossomos) localizados no centro das fibras musculares, em vez de na periferia. Esse achado na biópsia muscular também foi identificado em diversas outras doenças neuromusculares genéticas, incluindo miopatias relacionadas aos genes MTM1, BIN1 e DNM2.
A Miopatia Congênita Centronuclear (CNM) pode causar fraqueza muscular em qualquer fase da vida, desde o nascimento até o início da idade adulta. Essa fraqueza muscular pode levar a atrasos no desenvolvimento motor (engatinhar ou andar) e pode ser progressivamente lenta. Alguns indivíduos afetados podem necessitar de cadeira de rodas logo na infância. Outros sintomas incluem problemas respiratórios de leves a graves, queda da pálpebra superior (ptose), fraqueza dos músculos faciais, anormalidades nos pés, palato ogival (céu da boca alto) e curvatura anormal da coluna vertebral (escoliose) .
Desproporção Congênita do Tipo de Fibra - Congenital Fiber - Type Disproportion (CFTD)
A Desproporção congênita do tipo de fibra muscular (CFTD) foi escrita pela primeira vez por Brooke e colaboradores em 1969, e é herdada de forma recessiva. Quando observada ao microscópio, o tecido muscular com CFTD apresenta fibras musculares do tipo 1 (contração lenta) que são consistentemente menores do que as fibras musculares do tipo 2 (contração rápida).
A Desproporção congênita do tipo de fibra muscular (CFTD) causa fraqueza muscular, particularmente nos músculos dos ombros, braços, quadris e coxas. A fraqueza também pode afetar os músculos faciais, os músculos extraoculares que controlam o movimento dos olhos (oftalmoplegia) e os músculos da pálpebra superior (ptose). Indivíduos com CFTD geralmente apresentam rosto alongado, palato ogival e dentes apinhados. Os indivíduos afetados podem apresentar deformidades articulares (contraturas) e uma curvatura anormal da região lombar (lordose) ou uma curvatura lateral da coluna vertebral (escoliose). Aproximadamente 30% das pessoas com CFTD apresentam problemas respiratórios leves a graves relacionados à fraqueza dos músculos necessários para a respiração. Algumas pessoas que apresentam esses problemas respiratórios necessitam do uso de um aparelho para auxiliar na regulação da respiração à noite e, ocasionalmente, também durante o dia. Cerca de 30% dos indivíduos afetados têm dificuldade para engolir devido à fraqueza muscular na garganta. Raramente, pessoas com essa condição apresentam enfraquecimento e aumento do músculo cardíaco (cardiomiopatia dilatada). Variantes genéticas causadoras de CFTD também foram identificadas em outros genes, como ACTA1, TPM3 e SELENON.
Suscetibilidade a Hipertermia Maligna - Malignant Hyperthermia Susceptibility (MH)
Indivíduos sensíveis à Hipertermia Maligna (MH) podem experimentar uma vida diária normal sem qualquer sintoma ou fraqueza muscular. No entanto, quando expostos a certos agentes anestésicos, os pacientes podem experimentar um episódio de Hipertermia Maligna. A Hipertermia Maligna caracteriza-se por um estado hipermetabólico, causando um aumento anormal do calor, com rigidez excessiva e quebra muscular associada, e aumento da frequência cardíaca. As complicações graves da HM incluem: lesão cerebral, sangramento interno, parada cardíaca e/ou falência de múltiplos órgãos. As complicações cardiovasculares associadas podem ser fatais. Qualquer indivíduo com uma mutação(ões) do RYR-1, se a anestesia for necessária para um procedimento médico-cirúrgico, é aconselhado a tomar "precauções contra hipertermia maligna". Indivíduos com mutações RYR-1 suscetíveis a MH também correm risco de rabdomiólise, bem como outras dores e cãibras musculares relacionadas ao calor e ao esforço. Rabdomiólise é o termo geral para ruptura muscular associada a uma ampla variedade de gatilhos externos, incluindo: exercícios extenuantes além do limite de fadiga, abuso de drogas ou álcool, uso de suplementos ou certos medicamentos, doença viral recente ou trauma muscular. Os sinais e sintomas da rabdomiólise incluem dor muscular intensa, elevação repentina e queda subseqüente dos níveis séricos de creatina fosfoquinase (CPK) e produtos da quebra muscular na urina ("mioglobinúria"). O curso da rabdomiólise é caracterizado principalmente por mialgia (dor muscular) com aumentos leves a moderados da CPK. Nesses casos leves, muitos indivíduos não procuram atendimento médico. No entanto, em alguns o curso clínico é grave, resultando em hiper-CK-emia profunda, insuficiência renal aguda, síndrome do compartimento, coagulação intravascular disseminada, arritmias cardíacas secundárias a desequilíbrios eletrolíticos e, possivelmente, parada cardíaca se não tratada. Portanto, indivíduos com mutações no RYR-1, especialmente aquelas conhecidas por estarem associadas à suscetibilidade ao HM, devem estar cientes dos gatilhos da rabdomiólise e podem querer consultar um médico antes do início de um regime de exercícios e/ou esportes. Pode haver um papel do dantroleno como agente profilático na prevenção da rabdomiólise e de outros sintomas musculares relacionados ao esforço e ao calor.

