O estilo de vida levado por um indivíduo afetado por uma miopatia congênita é fundamental para o controle dos sintomas da condição, embora não existam tratamentos específicos para a cura da doença. Essas intervenções são consideradas tratamentos de suporte e visam preservar a função muscular, maximizar a independência, e melhorar a qualidade de vida do paciente.
O tratamento da miopatia congênita deve ter uma abordagem multidisciplinar e individualizada, focada na reabilitação e no suporte contínuo para otimizar a funcionalidade e o bem-estar. Esse estilo de vida a que me refiro, seria sustentado por um tripé de condutas, sintetizado no Controle de Stress, Nutrição, e Atividade Física.
 Controle do Stress
Controle do Stress
O controle do stress (stress management) é de extrema importância para a manutenção da saúde e qualidade de vida de indivíduos portadores de uma miopatia congênita. Embora não haja a cura ou tratamento específico para a doença em si, muitas vezes a única terapia indicada é de suporte, como fisioterapia.
Contudo, existem outras condutas que podem ser tomadas visando a mitigação dos sintomas da doença, melhoria na qualidade de vida dos indivíduos, e que inclusive interferem positivamente no prognóstico da doença. Essa conduta diz respeito a uma série de mudança ou incorporação de hábitos vida, chamada de Controle de Stress, e que é composta em vários fatores interligados:
 Nutrição
Nutrição
A qualidade de vida de um indivíduo afetado por uma miopatia é fortemente influenciada pela capacidade de manter a funcionalidade e a autonomia o máximo possível, apesar da fraqueza muscular, que é o sintoma mais comum. Neste contexto, a importância do controle nutricional é crítica e multifatorial, sendo uma parte essencial para seu tratamento de suporte.
No caso específico da Miopatia Centronuclear, por ser uma doença neuromuscular que causa fraqueza muscular significativa (hipotonia), pode levar a várias complicações que exigem um acompanhamento nutricional individualizado e rigoroso, fundamental para minimizar a perda de massa muscular, garantir o aporte energético adequado, e prevenir complicações secundárias à fraqueza. Esse manejo nutricional não visa curar a doença, mas sim otimizar a saúde geral, apoiar a função muscular e respiratória, e prevenir complicações que impactam severamente a qualidade de vida.
Não existe uma dieta ou protocolo de suplementação único para todos os indivíduos com miopatia congênita. A dieta e suplementação deve garantir um aporte energético suficiente para as necessidades metabólicas e para preservar a já tão sofrida massa muscular. O cálculo do gasto energético total precisa ser individualizado, considerando o nível de atividade física e a gravidade da miopatia.
O plano deve ser altamente individualizado e baseado na avaliação clínica, estado nutricional atual e exames laboratoriais. Para garantir segurança e eficácia, as intervenções nutricionais e a suplementação devem ser sempre orientadas por um médico e um nutricionista especializados ou com conhecimento em doenças neuromusculares. Esse planejamento nutricional se baseia em algumas premissas básicas:
Em resumo, uma nutrição bem planejada e a suplementação direcionada são ferramentas de suporte poderosas que visam otimizar a função muscular residual, prevenir deficiências nutricionais, apoiar a saúde óssea e melhorar a qualidade de vida do indivíduo com miopatia congênita.
 Atividade Física
Atividade Física
Sempre digo que a atividade física, no contexto das miopatias congênitas, vai além da função de mero suporte clínico, pois ela se estabelece como um elemento fundamental na promoção da qualidade de vida. A atividade física é crucial não apenas para retardar o declínio funcional, mas também para promover o bem-estar psicossocial do indivíduo afetado pela doença.
Para indivíduos com miopatia congênita, a atividade física é uma ferramenta terapêutica que busca promover a capacidade do indivíduo agir de forma mais autônoma possível, além de atuar na conexão social e dignidade pessoal, situações estas tão importantes quanto qualquer intervenção médica para alcançar uma qualidade de vida.
A atividade física capacita o indivíduo, dando-lhe um sentimento de propósito, controle e autoestima, que são um pilares da qualidade de vida.
A Declaração de Consenso sobre Padrão de Cuidados para Miopatias Congênitas, publicada em 2012 no Journal of Child Neurology, tratou a atividade física no contexto de se enfatizar a importância de um programa estruturado, mas altamente individualizado, para manter a função e a mobilidade.
O consenso reconheceu que, embora não exista cura, a fisioterapia e a terapia ocupacional (TO) são cruciais para o manejo da doença, e como promoção da qualidade de vida do indivíduo.
As diretrizes do consenso de 2012 sobre atividade física e reabilitação se referiam principalmente aos seguintes pontos:
Em essência, a declaração de consenso enfatizou que a atividade física para miopatias congênitas não é uma atividade de "ganho de força" típica, mas sim uma intervenção de manutenção e reabilitação focada em preservar a função e evitar as complicações secundárias da fraqueza, mas sobretudo e principalmente como agente de promoção de qualidade de vida.
Com os recentes avanços da ciência genética, a grande maioria dos diagnósticos de uma miopatia causada pela mutação no gene RYR1 acontece nos primeiros anos de vida até a adolescência. Entendo que foi pensando nesse público, que a Fundação RYR-1 @theryr1foundation resolveu criar um elemento que de alguma forma comunicasse com esse público infanto-juvenil, o qual é uma parte considerável da comunidade RYR1.
E neste contexto que foi apresentado à comunidade RYR1, durante a Conferência Internacional de Famílias, realizada em julho deste ano em Pittsburgh, E.U.A., o Ryan, o novo mascote da Fundação RYR1, o qual foi calorosamente bem recebido por toda comunidade RYR1.
A fundação informou que a partir de agora, Ryan será sempre apresentado em boletins informativos, mídias sociais e site da organização, envolvendo-se em uma variedade de atividades. Ryan toca piano, pinta e participa de esportes, incluindo boliche adaptativo, passeios a cavalo, hóquei e muito mais.
O nome "RYAN" é um aceno para o receptor de ryanodina, que é codificado pelo gene RYR1, gene responsável pelas doenças relacionadas ao RYR1. Ryan é retratado segurando um dedo para cima para representar o número "1,", enquanto as bochechas vermelhas do personagem simbolizam sensibilidade ao calor, um sintoma comum entre os membros da comunidade RYR1.
O conceito de mascote foi trazido à vida por voluntários dedicados da comunidade RYR-1-RD, incluindo integrantes do conselho administrativo e consultivo, e pelos pais Macie Soler-Sala e AJ Warren, ambos diretores criativos da Weiden+Kennedy e Goodby Silverstein & Partners, respectivamente, os quais desempenharam um papel fundamental no desenvolvimento de Ryan.
Os desenhos de Ryan foram criados por Chris Griarusso, um reconhecido artista e escritor indicado ao prêmio Harvey conhecido pela série G-Man na Image Comics e Mini Marvels para a Marvel Comics.
Ryan ganhou também uma música com um tema cativante, “Strength in Numbers”, que⇓ quer dizer fortes em número ou quantidade, ou tipo assim, a união é que faz a força, que foi escrita e interpretada por Macie Soler-Sala.
⇓ letra da música ⇓
♫ ♫ ♫ S-t-r-o-n-g (F-o-r-t-e)
Find out what it means to me (Descubra o que isso significa para mim)
Hope is in our DNA (A esperança está no nosso DNA)
We walk, we wheel, we skate, we play (Nós andamos, nós rodamos, nós patinamos, nós brincamos)
You got my back and I’ve got yours (Você está comigo e eu estou com você)
And every day we’re stronger than we were before (E cada dia somos mais fortes do que éramos antes)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
We got strength in numbers, you and me (Nós temos força em número, você e eu)
We never get tired of trying our best (Nunca nos cansamos de tentar o nosso melhor)
Until we have a cure, we just won’t rest (Até que tenhamos uma cura, não descansaremos)
So let’s raise our voices, let’s shout it out loud (Então vamos levantar nossas vozes, vamos gritar bem alto)
Every step forward is a reason to be proud (Cada passo à frente é motivo de orgulho)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
We got strength in numbers, you and me (Nós temos força em número, você e eu)
Together we’re better, together we’re stronger (Juntos somos melhores, juntos somos mais fortes)
What makes us different, makes us belong (O que nos torna diferentes, nos faz pertencer “ser aceitos”)
One, two, three, four (Um, dois, três, quatro..)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
We got strength in numbers, you and me (Nós temos força em número, você e eu)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
All I have on ain’t got nothing on me (Tudo o que tenho não tem nada contra mim)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
We got strength in numbers, you and me (Nós temos força em número, você e eu) ♫♫♫
"Estamos empolgados em receber oficialmente Ryan na comunidade RYR1", disse a Diretora Executiva, Ligação com Pacientes e Cofundadora, Lindsay Goldberg. "Muitos na comunidade podem se identificar com diferentes aspectos de Ryan, que incorpora o espírito de ser ativo, engajado e resiliente. Você verá mais do Ryan em breve!"
Desde que nasci, eu fui desafiado pelos sintomas de uma doença que me fazia ser diferente de outras crianças, ela me confrontava a todo tempo, me causava limitações físicas funcionais progressivas, e assim, causava a mim e à minha família um turbilhão de questionamentos e dúvidas sobre do que se tratava. Diante dos diagnósticos e prognósticos que recebia, dos mais piores possíveis, assim como pela falta de uma solução com um possível tratamento, eu, com toda minha inocência e intuito, resolvi que a melhor coisa que tinha a fazer era viver, e viver com intensidade, buscando a superação das minhas limitações, potencializando assim minha capacidade com o que tinha de físico, emocional, e intelectual. E sempre digo que apesar de tudo, eu nunca me vi no espelho como uma pessoa diferente, e neste contexto de vida, brinquei, namorei, me formei, casei, trabalhei muito, tive dois filhos, enfim, enquanto isso o mundo foi girando, a ciência descobrindo e explicando as coisas, até que quando tinha 44 anos, obtive meu diagnóstico sobre minha doença, ou seja, que tinha uma mutação no gene RYR1, e que me causava uma doença chamada de Miopatia Congênita Centronuclear, e que isso é que era a responsável pela condição que vivia desde que nasci. Pois bem, não mudou nada na minha vida, a não ser que descobri o endereço e nome da minha doença, o resto foi que ela era uma "Doença Rara", incurável e sem tratamento, e por fim, que pouco se conhecia sobre ela no meio médico-científico.
Sempre fui uma pessoa muito resiliente, que não só tem a força interior para enfrentar a realidade, por mais dura que seja, mas também tem a ambição intelectual e pessoal para moldar ativamente minha realidade em uma vida melhor, ou como diz minha mãe, mais "suave". Sempre fui uma pessoa da mente muito aberta ao conhecimento, ao questionamento, à observação, ao aprendizado, e essas minhas características não me fizeram ser uma pessoa acomodada. Não saber a princípio qual era doença que me acometia, e posteriormente descobri-la, e o pior que não tinha cura ou tratamento, nunca me causou sentimentos negativos, muito pelo contrário, me impulsionou a buscar viver melhor, mas também ao conhecimento.
essas minhas características não me fizeram ser uma pessoa acomodada. Não saber a princípio qual era doença que me acometia, e posteriormente descobri-la, e o pior que não tinha cura ou tratamento, nunca me causou sentimentos negativos, muito pelo contrário, me impulsionou a buscar viver melhor, mas também ao conhecimento.
A busca pelo conhecimento teórico sobre o mecanismo de funcionamento do gene, o RYR1, e como ele interferia no meu corpo foi um grande marco para meu conhecimento intelectual, assim como satisfação interna para minhas ansiedades. Contudo, o conhecer de perto, praticamente quase podendo o “tocar”, foi uma de minhas maiores experiências que já tive nesta minha peregrinação em busca de saber sobre a doença que me acomete desde que nasci. E isso se deu, depois de muito tempo, especificamente, recentemente, na última Conferência de Família e Workshop de Pesquisa do RYR1, realizada em julho de 2025 em Pittsburgh, EUA, durante uma reunião particular com o Dr. Filip Van Petegem. Na oportunidade, pude assistir um vídeo com a reconstituição em 3D “do meu RYR1”, ao mesmo tempo que o escutava fazer uma explanação específica sobre minha mutação. Dr. Van Petegem lidera um laboratório de pesquisa no Departamento de Bioquímica e Biologia Molecular na Universidade da Colúmbia Britânica (UBC), Vancouver, Canadá, no qual utiliza a cristalografia de raios X e microscopia crioeletrônica para estudar suas estruturas 3D, o que proporciona estudar a estrutura e função dos canais iônicos, com foco no músculo cardíaco e esquelético, e isso inclui o Receptor de Ryanodine (RyR). Um dos trabalhos do Dr Petegem consiste na importante abordagem em determinar as estruturas tridimensionais muito detalhadas desses canais do RYR1, permitindo a análise dos efeitos diretos das mutações na estrutura, e consequente interferência física no indivíduo afetado.
A título ilustrativo e como exemplo, gostaria de compartilhar com o leitor dessa postagem um pequeno vídeo criado pelo Dr Van Petegem, a partir da técnica acima descrita, da variante do meu RYR1 com a respectiva mutação genética. O vídeo lhe permitirá entender melhor a complexidade da estrutura molecular do gene RYR1, descrita na postagem anterior, O GIGANTE DO GENOMA QUE FAZ OS MÚSCULOS SE MOVEREM, e que neste caso se trata do meu RYR1.
O Vídeo
Para orientar um pouco melhor a apresentação deste vídeo, e assim entender o que acontece com meu gene RYR1, veja a seguir algumas da partes importantes da apresentação:
1). cada esfera que você vê representa um átomo individual;
2). o vídeo começa com uma visão 'top', ou seja, por cima do RYR1. Olhando no fundo da parte central, está a região chamada de 'poro', onde os íons de cálcio podem passar;
3). o vídeo amplia a localização da minha variante, E1175K. O aminoácido do 'tipo selvagem' é o chamado 'E', também conhecido como 'Glu' ou 'Glutamato'. Este aminoácido tem a propriedade especial de carregar carga negativa. No vídeo, este E1175 é mostrado em vermelho;
4). pode-se ver que o E1175 está apontando para outro aminoácido em uma cor diferente, em azul. Esse aminoácido, conhecido como 'R' ou 'Arg' ou 'Arginina', é especial porque carrega uma carga positiva. Assim, em pessoas sem nenhuma variante RYR1, haverá normalmente uma atração entre o 'E' carregado negativamente em vermelho, e o 'R' carregado positivamente em azul;
5). na variante demonstrada (a minha no caso), o E1175 carregado negativamente é substituído por 'K'. Este aminoácido, também conhecido como 'Lys' ou 'Lysine', também carrega carga positiva. Portanto, a variante não apenas abole a interação positivo-negativa normal, mas também coloca um aminoácido carregado positivamente (K) ao lado de outro (R), e isso desestabiliza a proteína. Por causa disso, o RYR1 se torna mais móvel e o poro pode abrir mais facilmente, ou seja, não funcionar da maneira correta. Esse defeito leva a um 'vazamento' de cálcio, que pode danificar o funcionamento normal dos músculos.
(VIDEO 1)
(VIDEO 2)
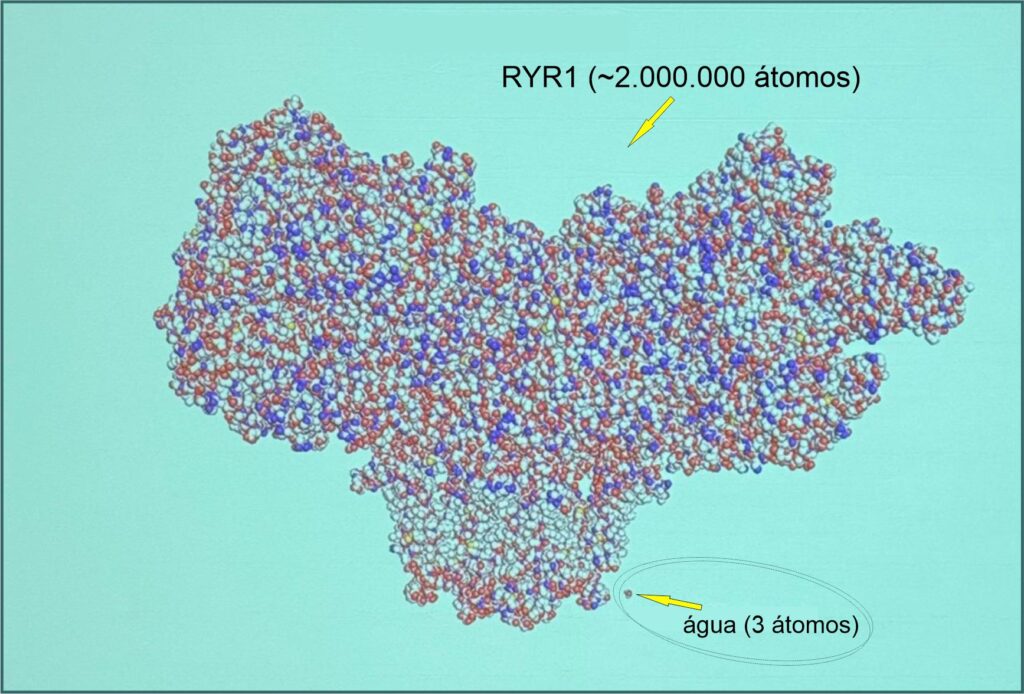
O RYR1 é um dos maiores genes do corpo humano, e é responsável para o funcionamento dos nossos músculos.
O gene RYR1 (Receptor de Rianodina 1) é um dos maiores e mais complexos genes do genoma humano. Ele contém mais de 100 exons e cerca de 15 mil pares de bases na sua sequencia de DNA, abrangendo uma grande extensão no cromossomo 19. A estimativa é que o gene RYR1 tenha cerca de 2 milhões de átomos, e esse é somente um cálculo de aproximação, pois a quantidade exata pode variar dependendo da sequência específica de nucleotídeos.
Para se ter uma idéia da complexidade do RYR1, ele é imensamente maior e mais complexo do que moléculas simples como da água, tão crucial para nossa vida, e a título de comparação, pasmem ! …o RYR1 tem 2 milhões de átomos, e a água, também conhecida como H20, tem somente 3 átomos.
O gene RYR1 que é crucial para a função do receptor de rianodina, que é um canal de cálcio localizado no Retículo 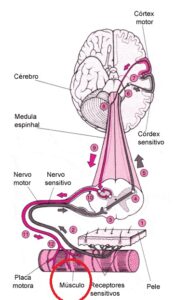 Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+
Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+) do RS para o citoplasma; 4). O cálcio dispara a contração muscular ao permitir a interação Actina-Miosina; 5). Para o relaxamento, o sinal cessa, o RYR1 se fecha, e rapidamente retorna o cálcio do citoplasma, devolvendo-o ao RS.
Em resumo, o RYR1 atua como o principal portão de saída do cálcio do reservatório intracelular. Ele é o mediador essencial que garante que o comando elétrico do nervo seja traduzido em movimento mecânico, controlando a entrada abrupta de cálcio que inicia a contração e, consequentemente, permitindo que a sua reabsorção finalize o processo para o relaxamento.
Como disse no início deste texto, devido ao tamanho e complexidade do RYR1, mutações nesse gene podem estar associadas a várias doenças musculares, incluindo:
O estudo científico do RYR1 é importante para entender essas condições ou doenças, e desenvolver tratamentos adequados em busca de minimizar seus efeitos e sintomas, pensando até na sua própria cura. Além disso, a análise genética pode ajudar em diagnósticos precoces e na prevenção de complicações associadas a essas doenças.
Durante o tempo que tenho acompanhado a movimentação de informações em torno das questões relacionadas ao RYR1, observo um grande esforço da comunidade científica em busca de entender o mecanismo de funcionamento deste complexo e grandioso gene, grande no tamanho, mas sobretudo grande em sua importância. Essas questões fazem com que o desafio ainda seja maior não só por conta dos indivíduos que têm uma doença a ele relacionada, mas também para compreensão sobre o impacto do envelhecimento celular em qualquer outro indivíduo. Observo também um grande movimento de trabalhos sendo desenvolvidos por cientistas de toda parte do mundo em busca de desenvolver uma droga para tratamentos adequados, para de repente ao menos minimizar seus efeitos e sintomas, até sua cura das doenças relacionadas ao RYR1. Particularmente, minha percepção é que as pesquisas através da técnica de reposicionamento de drogas, também chamado de redirecionamento de fármacos têm se mostrado mais promissoras no curto prazo. O “reposicionamento de drogas” se trata de uma estratégia que busca descobrir novas aplicações terapêuticas para medicamentos já existentes, que originalmente foram desenvolvidos para tratar outras doenças. Essa técnica tem a vantagem por seus reduzidos custos, assim como pela redução de tempo gasto no processo de desenvolvimento, e menores riscos no uso da eventual droga, pois já se conhece a farmacocinética e toxicidade do medicamento pesquisado, um exemplo real e prático de pesquisa em curso está se dando com o Sulfato de Salbutamol.
Contudo, a terapia genética também tem se mostrado uma abordagem promissora para o tratamento e cura de diversas doenças genéticas, oferecendo a possibilidade de corrigir ou substituir genes defeituosos. Embora este tipo de tratamento ainda esteja em desenvolvimento e não seja uma solução universal para algumas doenças, como por exemplo certas formas de miopatias ou doenças hereditárias, os resultados das pesquisas têm sido encorajadores. No entanto, a eficácia e a segurança da terapia genética podem variar dependendo da doença, do tipo de terapia utilizada, e condições do paciente. Além disso, questões éticas e de custo também são consideradas importantes. Contudo, enquanto a terapia genética oferece uma expectativa otimista, é uma área em evolução que requer mais pesquisas, altos recursos financeiros, e testes clínicos para se consolidar como a melhor solução para todas as doenças genéticas.
No caso específico das doenças relacionadas à mutação do gene RYR1, a terapia genética apresenta vários desafios, principalmente, como escrito no início deste texto, devido ao tamanho do gene, complexidade das doenças relacionadas a ele, mas também por considerar que os músculos representam aproximadamente 40% da massa total do corpo humano, e a maior parte dessa massa muscular é composta por músculos esqueléticos. Observe a seguir algumas das dificuldades:
Esses desafios tornam a pesquisa e o desenvolvimento de terapias para mutações no gene RYR1 complexos e exigem abordagens inovadoras e multidisciplinares.
Eu, enquanto portador de uma doença causada pela mutação nesse “grandioso” RYR1, tenho uma relação muito particular com esse gene, e a cada dia que passa o conheço um pouco mais, ..... confira minha próxima postagem intitulada “Eu vi o meu gene em 3D e entendi o que acontece dentro de mim”.
O SORRYR-1 mais uma vez cumpriu com seu objetivo de informar sobre as doenças relacionadas ao RYR-1 aos indivíduos afetados pela doença e seus familiares, profissionais da área da saúde, além de apoiar e participar de pesquisas científicas. Assim como já foi relatado nas redes sociais, eu fui convidado pela Dra Lizan Stinissen, do departamento de neurologia da Radboud University Medical Center, de Nijmegen, Holanda, para colaborar na elaboração de uma pesquisa internacional dirigida a pacientes com doenças relacionadas ao RYR1 visando sua participação em ensaios clínicos. O trabalho de elaboração da pesquisa foi concluído, pesquisa aplicada, e seu resultado foi apresentado pela Dra Lizan durante a Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos, tendo no final da apresentação, o orgulho de ouvir palavras de reconhecimento pela nossa participação e contribuição. O estudo se concentrou no que as pessoas com Miopatias Congênitas RYR1 e seus familiares esperam de futuros ensaios clínicos, e o que os incentivaria a participar dessas pesquisas. O objetivo deste estudo é fornecer aos pesquisadores clínicos, às empresas farmacêuticas, e à comunidade de indivíduos afetados pelas doenças relacionadas ao RYR-1, subsídios ao desenvolvimento de pesquisa em ensaios clínicos que sejam significativas e de importância terapêutica para os pacientes. O resultado da pesquisa fornecerá também aos profissionais de saúde uma visão sobre os sintomas mais importantes para os pacientes e as lacunas nas terapias disponíveis para os indivíduos com miopatia relacionada ao RYR1.
Medical Center, de Nijmegen, Holanda, para colaborar na elaboração de uma pesquisa internacional dirigida a pacientes com doenças relacionadas ao RYR1 visando sua participação em ensaios clínicos. O trabalho de elaboração da pesquisa foi concluído, pesquisa aplicada, e seu resultado foi apresentado pela Dra Lizan durante a Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos, tendo no final da apresentação, o orgulho de ouvir palavras de reconhecimento pela nossa participação e contribuição. O estudo se concentrou no que as pessoas com Miopatias Congênitas RYR1 e seus familiares esperam de futuros ensaios clínicos, e o que os incentivaria a participar dessas pesquisas. O objetivo deste estudo é fornecer aos pesquisadores clínicos, às empresas farmacêuticas, e à comunidade de indivíduos afetados pelas doenças relacionadas ao RYR-1, subsídios ao desenvolvimento de pesquisa em ensaios clínicos que sejam significativas e de importância terapêutica para os pacientes. O resultado da pesquisa fornecerá também aos profissionais de saúde uma visão sobre os sintomas mais importantes para os pacientes e as lacunas nas terapias disponíveis para os indivíduos com miopatia relacionada ao RYR1.
Veja a seguir como tudo se deu...
Pesquisa com Paciente com Doença Relacionada ao RYR1
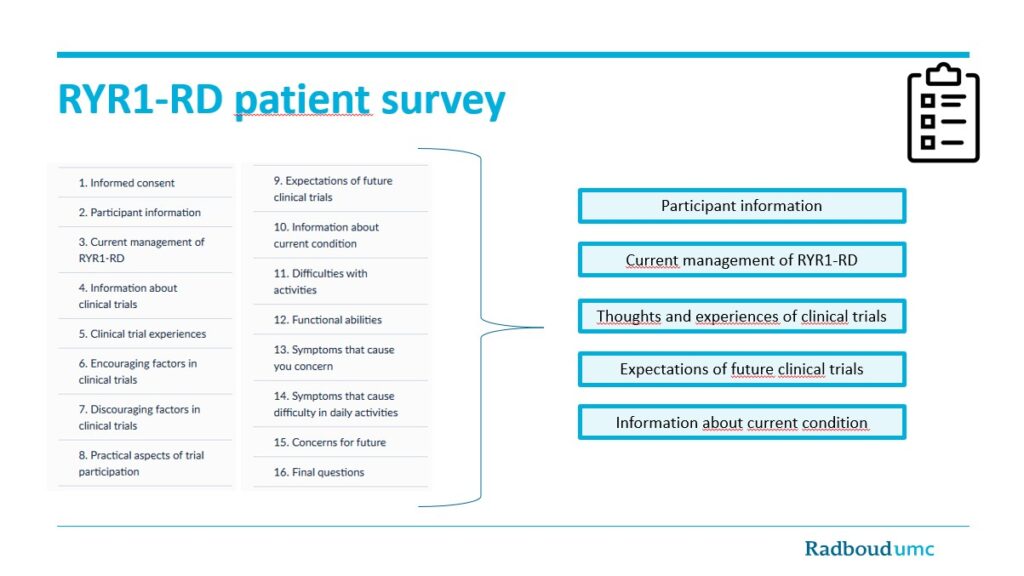
Este estudo se concentra sobre as preferências de ensaios clínicos de indivíduos com doenças relacionadas ao RYR1 (RYR1-RD).
Antecedentes e Objetivo do Estudo
Atualmente, existem vários estudos pré-clínicos e estudos de história natural em andamento sobre o RYR1-RD, assim como já houve dois ensaios clínicos até agora. Os possíveis próximos ensaios clínicos que estão por vir indicam ter bons resultados. A comunidade de pacientes pode contribuir para esses futuros ensaios apontando suas preferências e expectativas. Espera-se que este estudo possa fornecer aos pesquisadores clínicos, empresas farmacêuticas, e à comunidade de pacientes informações de importância significativas para pesquisas terapêuticas aos pacientes. O estudo destaca os maiores sintomas e queixas, buscando identificar medidas de relevantes resultado.
Este estudo é uma continuação de uma publicação sobre uma pesquisa online com pacientes iniciada pela Fundação RYR-1, combinada com depoimentos de pacientes apresentados durante o Workshop Internacional de Pesquisa realizado em 2022. O relatório completo do estudo anterior intitulada como: “Indivíduos e Famílias Afetados por Doenças Relacionadas ao RYR1: A Perspectiva do Paciente/Cuidador”, pode ser encontrado disponível no seguinte link aqui: https://ryr1.org/medical-literature/individuals-and-families-affected-by-ryr1-related-diseases-the-patient-caregiver-perspective
Como o Estudo Foi Estruturado
A pesquisa com pacientes RYR1-RD esteve disponível online entre 10 de março e 5 de maio de 2025. Ela incluiu perguntas sobre a condição dos participantes e como ela estava sendo tratada na época, além de suas opiniões e expectativas em relação às pesquisas e ensaios clínicos. A pesquisa foi disponibilizada em 6 idiomas: inglês, espanhol, português, francês, alemão e holandês. Como apoio ao processo de tradução e revisão da pesquisa, contamos com a colaboração de perto de membros da Fundação RYR-1 e palestrantes de acordo com o seu respectivo idioma, muitos dos quais eram também pacientes.
Primeiros Resultados e Próximos Passos
Os resultados fornecem informações sobre:
Um total de 152 pessoas, representando 19 países diferentes, responderam a pesquisa. Os resultados foram apresentados na Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos. No momento, estamos escrevendo um artigo científico com base nos resultados da pesquisa. Planejamos submetê-lo a uma revista médico-científico, para que tanto médicos quanto pesquisadores possam se beneficiar dos resultados. Esperamos que os resultados aumentem a compreensão dos pacientes sobre sua participação futura em pesquisas clínicas, assim como apoie os pesquisadores no desenvolvimento futuros de seus projetos de pesquisas clínicas. O artigo demonstrará a importância de refletir sobre as experiências, pensamentos e expectativas dos pacientes, e destaca a realização de um trabalho de cocriação com pesquisadores acadêmicos e a comunidade de pacientes. Uma atualização seguirá assim que o artigo for finalizado.
em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos. No momento, estamos escrevendo um artigo científico com base nos resultados da pesquisa. Planejamos submetê-lo a uma revista médico-científico, para que tanto médicos quanto pesquisadores possam se beneficiar dos resultados. Esperamos que os resultados aumentem a compreensão dos pacientes sobre sua participação futura em pesquisas clínicas, assim como apoie os pesquisadores no desenvolvimento futuros de seus projetos de pesquisas clínicas. O artigo demonstrará a importância de refletir sobre as experiências, pensamentos e expectativas dos pacientes, e destaca a realização de um trabalho de cocriação com pesquisadores acadêmicos e a comunidade de pacientes. Uma atualização seguirá assim que o artigo for finalizado.
RYR1-RD patient survey_LizanStinissen






O entendimento das pessoas evolui à medida que a ciência avança, e novas descobertas são feitas. Nesta dinâmica, o avanço médico e científico ao longo dos últimos anos teve um impacto significativo no diagnóstico e na percepção médica sobre as doenças.
Com o desenvolvimento de novas tecnologias, métodos de pesquisa e compreensão biológica, o conhecimento genético avançou de maneira notável, especialmente após se ter conseguido fazer o o mapeamento do genoma humano. Agora, através do resultado genético é possível não só diagnosticar uma doença, mas entender seu impacto no indivíduo, e adaptar as abordagens com base no perfil genético de cada paciente.
Nestes meus mais de 60 anos, senti na pele essa grande reviravolta na ciência, no que diz respeito ao conhecimento médico e diagnóstico da minha doença. Vivi até meus 44 anos, com vários diagnósticos errados, abordagens diferentes, até que tive finalmente o diagnóstico sobre a doença que me afeta, a Miopatia Congênita Centronuclear (MCC). Essa doença é causada pela mutação no gene RYR1, não tem cura, nem tratamento. Apesar da dureza sobre as informações sobre a doença, ter o diagnóstico de certa forma me trouxe uma “espécie de alívio“. O RYR1 é responsável pelo controle do fluxo de íons de cálcio para dentro da célula muscular, que faz o músculo contrair e relaxar.
Naquela época, o resultado histológico, feito por uma biópsia muscular, era tido como sendo uma informação importante no diagnóstico, e acompanhamento clínico, porque através dele podia-se saber e diferenciar se o indivíduo era afetado por exemplo, por Miopatia Congênita Centronuclear (MCN), Miopatia Central Core (MCC), Doença Multi-Minicore (DMm), ou Desproporção Congênita do Tipo de Fibra (DCTF). Aconteceu que através de minha vivencia em meio ao pequeno mundo dos afetados por essa doença, pude ver diferentes pessoas afetadas por MCN com a evolução diferente, assim como comparados com indivíduos em pior condições físicas em relação a um outro com MCC, ou DCTF, da mesma verificando também no inverso.
O avanço das técnicas de diagnóstico genético, nos trouxe uma mudança significativa, permitindo identificar diretamente variantes no gene RYR1, possibilitando uma visão mais precisa e específica da doença. Essa abordagem é preferível porque pode confirmar a presença de alterações no gene, mesmo na ausência de características histopatológicas distintivas, permitindo uma melhor orientação do prognóstico e o acompanhamento clínico do paciente.
Passado um tempo, descobri que no diagnóstico da doença, a histologia não tem um papel tão relevante quanto anteriormente, mas sim a herança e variação genética, em que é separado as formas recessivas e dominantes. Clinicamente observa-se evolução clínica com quadros mais graves em indivíduos com herança recessiva.
Em recente conversa com Michael F. Goldberg, MD, MPH, Presidente do Conselho, Co-Presidente de Pesquisa e Co-Fundador da @theryr1foundation, ele me disse: “Casos recessivos de RYR-1 tendem a ser mais graves quando associados a variantes do gene RYR1 resultando em expressão reduzida da proteína receptora RYR-1. As formas dominantes tendem a ser menos graves porque essas variantes geralmente não resultam em quantidades reduzidas da proteína receptora RYR-1. Os diagnósticos histopatológicos derivados da biópsia muscular (por exemplo, Centronuclear, Central Core, ou DCTF, etc.) não parecem ser muito informativos e não são específicos para fenótipos específicos mesmo para RYR1. Portanto, houve um afastamento do diagnóstico histopatológico e uma ênfase maior no diagnóstico genético .”
Em resumo, os diagnósticos histopatológicos obtidos a partir de biópsias musculares, como Centronuclear, Central Core, Desproporção Congênita do Tipo de Fibra, ou Multi Mini Core, não são muito informativos, e não são específicos para certos fenótipos. Isso levou a uma mudança para um maior enfoque no diagnóstico genético, que é mais preciso e informativo. Esse avanço no entendimento científico, permitiu a identificação e conhecimento das variantes genéticas, e sua relação com a gravidade das doenças associadas ao gene RYR1, crucial para um diagnóstico eficaz, e para o desenvolvimento de abordagens adequadas ao paciente.
A Miopatia Congênita Centronuclear é uma condição neuromuscular rara, com características de progressividade, e que pode levar a diversas complicações, incluindo problemas ortopédicos, como já relatado em postagem anterior (clique aqui), mas também podem ocorrer complicações respiratórios. As questões respiratórias podem surgir devido à fraqueza muscular que afeta os músculos respiratórios, resultando em dificuldades na ventilação e na troca gasosa. Para melhor entender o contexto desta questão das complicações respiratórias em indivíduos com miopatia relacionada ao RYR-1, o texto a seguir será dividido em tópicos conceituais e explanatórios.
OXIGÊNIO E O SISTEMA RESPIRATÓRIO
O oxigênio é de vital importância para o corpo humano, especificamente a cada célula, órgão e sistema do organismo. No caso dos músculos, o oxigênio desempenha um papel fundamental, como segue:
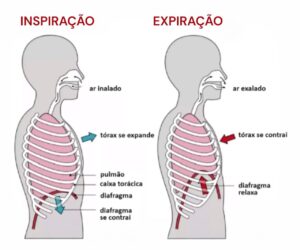 O sistema respiratório, através do processo de respiração, é o responsável por alimentar todo nosso oragnismo de oxigênio, elemento tão precioso e responsável por nos manter vivos. A respiração é um processo involuntário e automático que ocorre em duas etapas: Inspiração e Expiração.
O sistema respiratório, através do processo de respiração, é o responsável por alimentar todo nosso oragnismo de oxigênio, elemento tão precioso e responsável por nos manter vivos. A respiração é um processo involuntário e automático que ocorre em duas etapas: Inspiração e Expiração.
Trazendo toda essa questão da respiração para o contexto de um portador de uma miopatia, deve-se destacar que o diafragma é o principal músculo respiratório. O processo respiratório funciona assim:
RESPIRAÇÃO EM UM INDIVÍDUO COM MIOPATIA
Um indivíduo com miopatia tende ter fraqueza no diafragma e nos músculos abdominais, e essa situação pode dificultar a desobstrução das vias aéreas, pois se torna difícil inspirar profundamente e expirar de forma forte e completa. Se por exemplo um indivíduo com miopatia pega um resfriado, com seus músculos respiratórios enfraquecidos, ele pode desenvolver uma pneumonia. Esse processo inflamatório nos pulmões pode enfraquecer ainda mais os músculos e causar mais problemas na desobstrução das vias aéreas. A parede torácica e o abdômen formam a caixa torácica. Se a parede torácica estiver fraca, durante a inspiração, o tórax pode se movimentar para dentro em vez de se mover para fora. Isso dificulta ainda mais a respiração profunda. À medida que a parede torácica se enfraquece e se movimenta menos, ela também fica rígida. A rigidez da parede torácica torna ainda mais difícil a respiração profunda.
A fraqueza da caixa torácica também pode causar instabilidade da coluna. Isso pode causar uma curvatura na coluna, o que resulta em cifose ou "corcunda" nas costas. Essa instabilidade também pode causar a curvatura lateral da coluna, o que resulta em escoliose. Essas curvaturas podem limitar o movimento da parede torácica durante a respiração, o que dificulta a respiração profunda e resulta em menor volume dos pulmões. Juntos, esses problemas podem levar a uma situação em que os músculos respiratórios não funcionam bem o suficiente para trazer oxigênio para dentro do corpo e eliminar o dióxido de carbono. Isso se chama insuficiência respiratória.
COMPLICAÇÕES RESPIRATÓRIAS EM UM INDIVÍDUO COM MIOPATIA
Assim como ocorre com outros sintomas nos indivíduos afetados por uma das doenças relacionadas ao RYR-1, a gravidade das complicações respiratórias também varia de indivíduo para indivíduo. Assim, algumas pessoas com doenças relacionadas ao RYR-1 podem não ter problemas. Alguns podem ter problemas leves, mas precisam de auxílio para respirar durante o sono ou quando estão doentes. Em casos graves, alguém com doença relacionada ao RYR-1 pode precisar de ventilação mecânica para respirar. Segue algumas das complicações respiratórias que podem ocorrer incluem:
É importante que os indivíduos com Miopatia Congênita Centronuclear sejam monitorados de perto quanto à função respiratória e recebam cuidados multidisciplinares para gerenciar eventuais complicações. O diagnóstico preciso de complicações respiratórias envolve uma variedade de exames que permitem aos médicos avaliar a função pulmonar, iniciando por um exame clínico, raio-x, tomografia computadorizada, teste de função pulmonar (TFP), polissonografia, exames laboratoriais, dentre outros. Uma vez identificado uma complicação respiratória, o tratamento pode incluir fisioterapia respiratória, suporte ventilatório e acompanhamento regular com especialistas em pulmão. E caso o indivíduo, afetado por Miopatia Centronuclear pela mutação no RYR-1, necessite de hospitalização, é importante se ter preventivamente um plano de suporte respiratório e orientação clínica. Na necessidade de uma cirurgia, os médicos deverão adotar precauções para suporte respiratório antes e depois da cirurgia, além de serem alertados sobre a sussetibilidade à Hpertermia Maligna.
ABORDAGENS RESPIRATÓRIAS PREVENTIVA E TERAPEUTICA EM UM INDIVÍDUO COM MIOPATIA¹
Assim como relatado em postagem anterior Abordagem Respiratória na Miopatia Centronuclear (clique aqui) a fisioterapia respiratória é responsável pela minimização dos comprometimentos respiratórios nas miopatias centronucleares, pois interfere na progressão da perda muscular, minimiza as complicações da perda de capacidade pulmonar, mantem a funcionalidade respiratória como fala, deglutição e tosse. Fazem parte das estratégias utilizadas no tratamento:
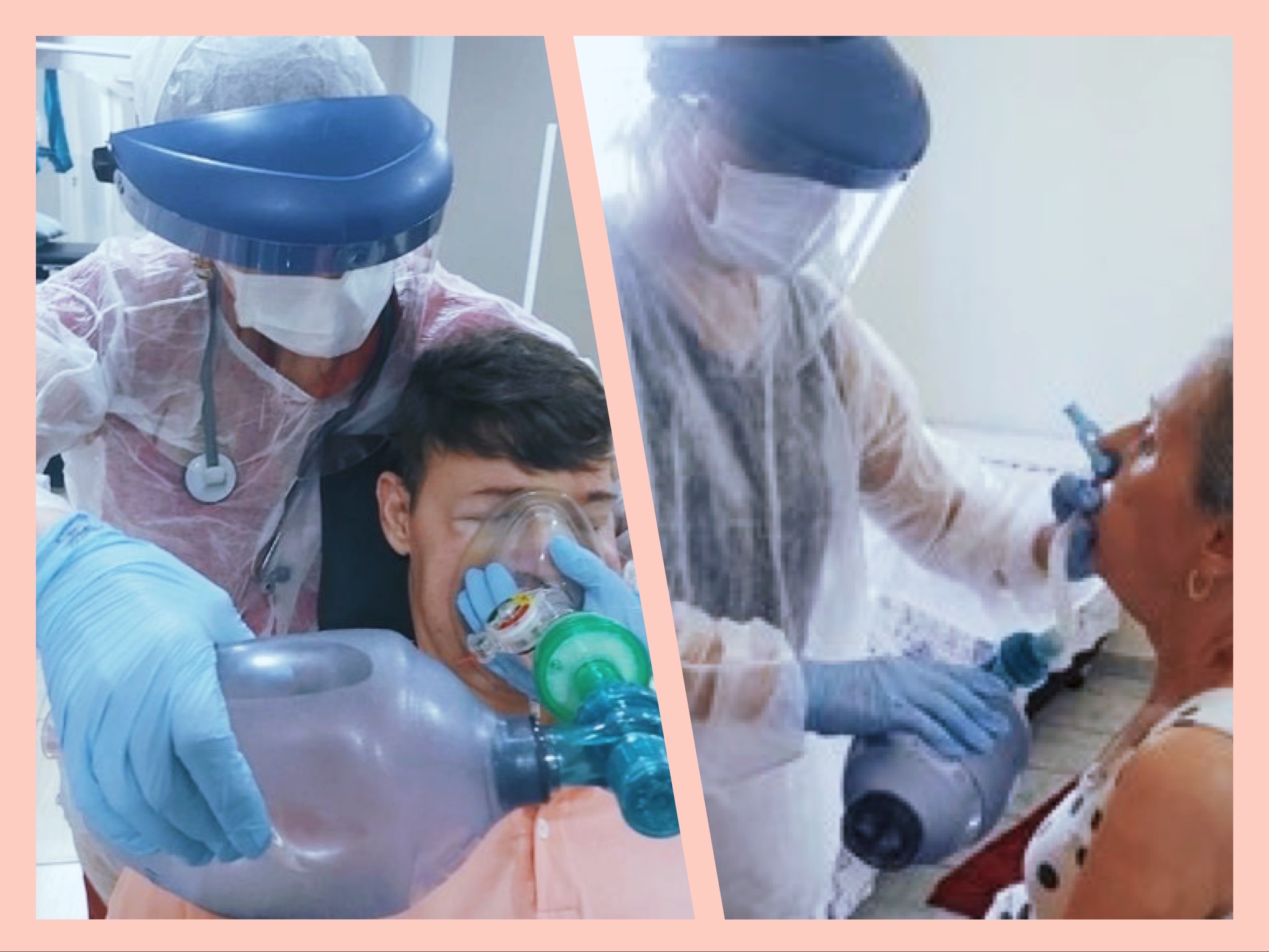
Fig 1 -Exercício de empilhamento de ar \ Fig 2 - Exercícios respiratórios para manutenção da capacidade pulmonar e treinamento da musculatura bulbar (ambos utilizando a bolsa de insuflação (ambu)
O cuidado respiratório deve ser uma prioridade nos portadores de Miopatia Centronucelar, e o profissional deve ser capaz de identificar a alteração funcional e correlacionar com a melhor terapia a ser aplicada, sendo esta a melhor estratégia para evitar maiores complicações para o paciente.
¹ texto cedido por colaboração da fisioterapeuta Alessandra Dorça do Instituto Alessandra Dorça @institutoaledorca
As doenças relacionadas ao RYR1 são identificadas com base em sua classificação histopatológica, isto é, pela aparência da biópsia do músculo na lâmina do microscópio. A diferenciação encontrada na biópsia designará o tipo da doença, se é por exemplo, Miopatia Central Core, Miopatia Multiminicore, Miopatia Centronuclear, ou Desproporção Congênita de Tipos de Fibras.
A Miopatia Congênita Centronuclear (MCCN) e a Miopatia Central Core (MCC) são ambas doenças musculares hereditárias causadas por mutações genéticas. A MCCN pode estar associada a mutações em diferentes genes, no DNM2, BIN1, MTM1, e RYR1, já a MCC está associada somente ao gene RYR1.
Esses tipos de doenças relacionadas ao RYR1 variam amplamente em termos dos seus diferentes sinais e sintomas, de quando eles inicialmente se apresentaram, além da sua respectiva gravidade. Embora sejam altamente variáveis, os sintomas presentes também dependem se a mutação do gene RYR1 é autossômica dominante ou autossômica recessiva.
Uma pergunta que sempre chega até mim é sobre as diferenças entre a Miopatia Centronuclear e a Miopatia Central Core. Assim, eu, enquanto portador da Miopatia Congênita Centronuclear (MCN), buscarei esclarecer pontualmente neste texto, me atendo ao tipo que me acomete, que é a pela mutação no gene RYR1.
Estas doenças apesar de terem sintomas parecidos e compartilharem de algumas características clínicas em comum, se confundem entre si, e apresentam com algumas diferenças distintas:
Miopatia Congênita Centronuclear (MCCN)
Miopatia Central Core (MCC)
Em resumo, a Miopatia Congênita Centronuclear e a Miopatia Central Core, ambas doenças relacionada ao RYR1, têm seus sintomas e sinais físicos que podem se parecer, podem se confundir, mas são diferentes, a contar da análise histológica das células musculares em uma biópsia, exame este que é crucial para diferenciar entre as duas condições e determinar o diagnóstico correto, conduta médica, tratamento, e até prognóstico de evolução.
Nota: todo o material escrito nesta página é oriundo de pesquisa científica, e conclusões próprias do autor deste website, que convive como portador, desde o nascimento, com a Miopatia Congênita Centronuclear causada pela mutação no gene RYR-1
ARMGO PHARMA PÚBLICA IMPORTANTES E POSITIVOS RESULTADOS DO ENSAIO DE FASE 1B DO RYCALl® ARM210 PARA O TRATAMENTO DE MIOPATIAS RELACIONADAS AO RECEPTOR DE RIANODINA 1
ARMGO Pharma, Inc. (ARMGO), uma empresa privada no setor biofarmacêutico que desenvolve uma nova classe de drogas de moléculas pequenas ou micro moléculas conhecidas como Rycals®, anunciou em 29 de janeiro de 2024 a publicação dos resultados de um estudo de Fase 1b de seu Rycal ARM210 (também conhecido como S48168), para o tratamento de miopatias relacionadas ao receptor 1 de Ryanodina (RYR1-RM), uma doença muscular órfã, também chamada de “doença rara”, por ser uma doença que afeta uma pequena percentagem da população.
2024 a publicação dos resultados de um estudo de Fase 1b de seu Rycal ARM210 (também conhecido como S48168), para o tratamento de miopatias relacionadas ao receptor 1 de Ryanodina (RYR1-RM), uma doença muscular órfã, também chamada de “doença rara”, por ser uma doença que afeta uma pequena percentagem da população.
Os dados foram publicados em um artigo intitulado 'Rycal S48168 (ARM210) para Miopatias Relacionadas ao RYR1: um ensaio de fase um, estudo em aberto, e de ensaio de escalonamento de dose', de autoria do Dr. Joshua Todd et al, no Journal eClinicalMedicine, parte da família de publicações Lancet. O artigo revisa os dados do estudo de Fase 1b do ARM210 e seu novo mecanismo de ação alostérico (MoA) visando a causa raiz da doença relacionada ao RYR-1 (RYR1-RM): mutação do Receptor 1 de Ryanodina (RYR1).
O gene RYR1 que codifica o Receptor 1 de Ryanodina RYR1, um canal intracelular de liberação de cálcio, vaza em doenças musculares. Vazamentos intracelulares de cálcio causados por canais RYR1 com mutação prejudicam a contração muscular, levando à fraqueza muscular e perda de função, e ativam vias tóxicas que danificam os músculos, causando os sintomas das doenças relacionadas ao RYR1.
O ensaio de Fase 1b, aberto, de escalonamento de dose confirmou a segurança, tolerabilidade e farmacocinética da dosagem de 120 e 200 mg de ARM210 diariamente durante 29 dias em homens e mulheres adultos afetados pelas doenças relacionadas ao RYR1 (RYR1-RM).
É importante ressaltar que o estudo também demonstrou eficácia preliminar no grupo de dose mais alta em dois sintomas característicos das doenças relacionadas ao RYR1 (RYR1-RM): 1) alívio significativo da fadiga avaliada pelo sistema PROMIS-fatigue (Patient-Reported-Outcome Measurement Information System) t-scores, e 2) melhora da força dos proximais avaliada pelo exame físico de abdução do ombro (Medical Research Council Grading). Estes resultados justificam o desenvolvimento futuro do ARM210 como um potencial tratamento e modificador da doença para as miopatias relacionadas ao RYR1 (RYR1-RM) em um ensaio de Fase 2 randomizado e controlado por placebo.
O ensaio de Fase 1b concluído foi conduzido em colaboração com o National Institute of Neurological Disorders and Stroke (NINDS) e National Institute of Health (NIH) sob um Acordo Cooperativo de Pesquisa e Desenvolvimento (CRADA - Cooperative Research and Development Agreement ), com o apoio da Fundação RYR-1, Pittsburgh, PA, EUA.

(Foto esquerda para direita: Dr. Tokunbor Lawal (autor da publicação), Dr. Mike Goldberg (co-fundador/Co-presidente de Pesquisas da Fundação RYR-1), e Dr. Payam Mohassel (Pesquisador Principal e Autor Senior)).
“Estamos muito satisfeitos com os resultados do ensaio com as doenças relacionadas ao RYR1 (RYR1-RM) conduzido em conjunto com o NIH, pois o estudo confirmou a segurança e tolerabilidade do ARM210, mas o mais importante, demonstrou pela primeira vez que o nosso Rycal®, ARM210, pode reverter os sintomas desta doença muscular crônica e devastadora em um curto período de tratamento. Isso é muito promissor”, disse Gene Marcantonio, M.D., Ph.D., CEO da ARMGO Pharma. “Esperamos, portanto, continuar rapidamente no desenvolvimento do ARM210 para levar este primeiro e potencial tratamento aos pacientes com as doenças relacionadas ao RYR1, com o apoio da Fundação RYR-1 e da comunidade de pacientes.”
Michael F. Goldberg, MD, MPH, co-presidente de pesquisa da Fundação RYR-1 acrescentou: “Estamos entusiasmados com a publicação deste importante estudo, pois representa um farol de esperança para muitos indivíduos e famílias de todo o mundo afetados pelas doenças relacionadas ao RYR1. Estamos ansiosos pelas próximas etapas de ensaios no desenvolvimento dessa importante droga.”
Mais informações sobre este estudo de Fase 1b podem ser encontradas online em: https://clinicaltrials.gov/study/NCT04141670. O ensaio foi apoiado pelos Programas de Pesquisa Intramural do NIH/NINDS, NIH/NINR, um NIH Clinical Center Bench to Bedside Award (2017-551673) e pelo parceiro de colaboração anterior da ARMGO, Les Laboratoires Servier. O conteúdo é de responsabilidade exclusiva dos autores e não representa necessariamente a opinião oficial dos Institutos Nacionais de Saúde.
BREVE REVISÃO SOBRE A FUNÇÃO DO RYR1 E DOENÇAS RELACIONADAS
Os RYRs são canais homotetraméricos de liberação de cálcio intracelular responsáveis pelo fluxo de Ca2+ dos retículos sarcoplasmáticos/endoplasmáticos (RS/RE) para o citoplasma da maioria dos tipos de células. RYR1 é a isoforma predominante no músculo esquelético de mamíferos, onde a liberação de Ca2+ via RYR1 é necessária para o acoplamento excitação-contração e função muscular normal. Já o RYR2 é a isoforma predominante no músculo cardíaco onde a liberação de Ca2+ via RYR2 é necessária para a função normal do músculo cardíaco. Mutações genéticas humanas nos genes RYR1 e RYR2 fazem com que o cálcio vaze dos canais RYR, levando à doença (Figura 1).
Os canais RYR normalmente alternam entre um estado de repouso (fechado) e excitado (aberto). Em certas doenças, em que existe a mutação genética, o RYR é modificado e torna-se incontinente ou seja, fica vazando. No caso do RYR1, este desempenha um papel crítico no músculo esquelético, e as mutações do RYR1 em humanos levam a uma miopatia progressiva, conhecida como doenças relacionadas ao RYR1 (RYR1-RM), privando o músculo da capacidade de responder eficazmente aos sinais de contração, levando à fraqueza muscular.
Sobre o Rycals® - A inibição do canal interromperia o vazamento, mas esta intervenção geralmente não seria benéfica, uma vez que bloquearia a função normal do RYR. O Rycals®, são moléculas que podem restaurar a função normal do canal sem bloquear o RYR, e abrem a possibilidade de intervenções terapêuticas.
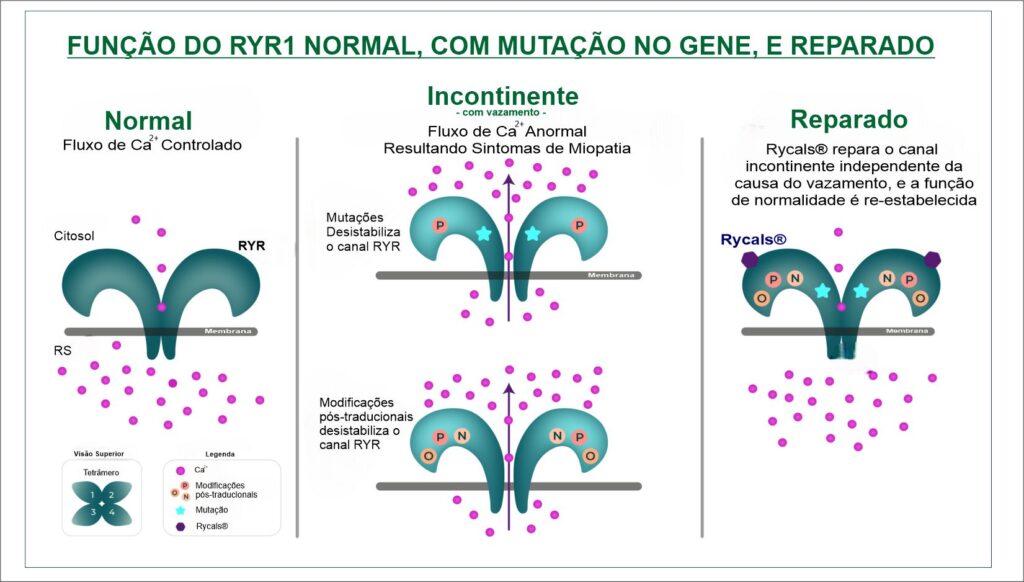
Figura 1: Modelo da função do RYR. O RYR controla o fluxo de cálcio (Ca2+) de dentro do retículo sarcoplasmático (RS) através da membrana até o citoplasma. (Quadro Esquerdo) O RYR normal regula o fluxo de cálcio alternando entre um estado fechado e aberto. (Quadro Meio) Na RYR1-RM, o RYR mutado apresenta vazamento, levando ao fluxo anormal de cálcio, resultando em sintomas da doença. Modificações pós-traducionais* (MPTs) do RYR agravam ainda mais o vazamento. Em outros distúrbios musculares, como DMD, insuficiência cardíaca ou sarcopenia, as modificações pós-traducionais (MPTs) do RYR podem causar vazamento no canal, levando a um fluxo anormal de cálcio, resultando em sintomas da doença. (Quadro Direito) Rycals® liga-se aos canais incontinentes (com vazamento) e repara o vazamento independentemente da causa do vazamento, restaurando a função normal do canal. O Rycals® não bloqueia o RYR.
*As modificações pós-traducionais (MPTs) são modificações químicas e estruturais de uma cadeia proteica após a sua tradução. Estas modificações podem determinar a atividade, a localização e interações com outras proteínas.

Em recente postagem na página do SorRYR-1 no Instagram (@sorryr_1) em que me caracterizei com pequenas frases, fui muito questionado sobre um termo utilizado nos EUA, #ambulatorywheelchairuser, mas que aqui no Brasil ainda não é muito conhecido, “Cadeirante Ambulatório”.
Indivíduos afetados por uma doença neuromuscular tem uma grande probabilidade de ter sua mobilidade limitada no decorrer do tempo, e essa situação pode evoluir de maneira gradativa ou repentinamente.
A maneira com que nós, pessoas com deficiência física (PCD), encaramos nossa situação e a forma que vivemos diante de nossas adversidades evoluíram muito nos últimos anos, assim como pela maneira que sociedade nos veem.
Para que essa evolução acontecesse alguns conceitos tiveram que ser inseridos em nosso dia a dia, como por exemplo a inclusão social, acessibilidade, capacitismo, enfim, mas alguns termos também tiveram que ser lançados para nos caracterizar melhor diante da sociedade, como por exemplo, “Cadeirante Ambulatório”.
Cadeirante Ambulatório refere-se a pessoas com deficiência física ou doença crônica que usam cadeira de rodas, embora possam ter alguma capacidade de andar em circunstâncias limitadas e particulares. Há muitas razões pelas quais uma pessoa pode ser um cadeirante ambulatório, sendo a principal para melhorar sua liberdade e a qualidade de vida. Outro termo importante destacar neste momento é que existe o cadeirante ativo e cadeirante passivo, sendo que o primeiro consegue por si mesmo tocar a rodas a cadeira de rodas, e o passivo depende outra pessoa.
Recentemente foi publicado um artigo médico-científico tratando sobre a Pancreatite em Indivíduos com Doenças Relacionadas ao RYR1. O estudo teve participação de pesquisadores do Reino Unido, Holanda e Estados Unidos, além do nobre apoio da Fundação RYR1.
Mutações no gene do receptor de rianodina do músculo esquelético (RYR1) são uma causa comum de doenças neuromusculares hereditárias e têm sido associadas a um amplo espectro de fenotípico, variando de várias miopatias congênitas de início precoce com fraqueza muitas vezes substancial, até fenótipos induzidos em indivíduos “normalmente fortes” tais como a Rabdomiólise por Esforço (ERM) e suscetibilidade aos efeitos relacionados à anestesia, como a Hipertermia Maligna (HM). O gene RYR1 codifica o principal canal de liberação de cálcio do retículo sarcoplasmático (SR) com um papel crucial
no acoplamento excitação-contração (ECC), processo pelo qual um impulso neuronal elétrico é traduzido em contração muscular por meio da liberação de cálcio intracelular, estimulando o filamento contrátil. Mutações no gene RYR1 associadas à fraqueza muscular permanente normalmente prejudicam o processo excitação-contração (ECC) de forma eficaz, enquanto aquelas associadas com ERM e MH resultam em um receptor RYR1 hiperexcitável e muitas vezes aumenta desproporcionalmente a liberação de cálcio.
Os genes RYR1s foram implicados em processos essenciais de sinalização de cálcio em uma ampla gama de tecidos, mas as manifestações de doenças humanas associadas ao mau funcionamento do RYR1 em outros órgãos além do músculo esquelético até agora receberam pouca atenção. Descobriu-se que os genes RYR1s são amplamente expressos no pâncreas de mamíferos, através de seu papel nos processos de sinalização de cálcio intracelular, e têm sido criticamente implicados na função pancreática endócrina e exócrina.
No estudo foi relatado três casos com características principais de um distúrbio relacionado ao RYR1 e uma história adicional de pancreatite (aguda) inexplicável, sugerindo uma nova associação clínica de função perturbada do RYR1 não relacionada ao músculo estriado. Os três pacientes com diagnóstico de Miopatia Central Core (CCD), Síndrome de King-Denborough (KDS) e Suscetibilidade à Hipertermia Maligna (MHS), respectivamente, que além de seu (suposto) distúrbio relacionado ao RYR1 também desenvolveram sintomas e sinais de pancreatite aguda. Em dois pacientes, os episódios foram recorrentes, com grave envolvimento multissistêmico e sequelas. A sinalização de cálcio mediada por RYR1 desempenha um papel importante na função pancreática normal, mas também tem sido criticamente implicada na fisiopatologia da pancreatite aguda, particularmente nas formas induzidas por ácidos biliares e etanol. Os resultados de modelos animais relevantes indicam que os danos pancreáticos nestas condições podem ser melhorados através da administração do antagonista específico de RYR1, dantroleno, e de outros compostos que modificam o metabolismo pancreático, incluindo a sinalização de cálcio. Estas observações sugerem que os pacientes com variantes de ganho de função do RYR1 podem ter um risco aumentado de desenvolver pancreatite aguda, uma condição que deve, portanto, ser considerada na vigilância da saúde desses indivíduos.
Saiba mais detalhes sobre o tema
deste texto lendo a publicação
completa no artigo científico
clicando na imagem ao lado ⇒
A terapia genética que era tida como uma futura grande promessa para o tratamento de miopatias relacionadas ao RYR1, se torna uma realidade com a publicação do recente relato científico da primeira correção por Edição Prime de uma mutação no gene RYR1.
A Fundação RYR-1 (https://ryr1.org/) cumprimentou a todos no início de ano com um “Feliz 2024, mas também compartilhou a informação que financiou uma pesquisa incrivelmente importante com o Dr. Jacques P. Tremblay, um pesquisador na Universidade Laval em Quebec. As descobertas e resultados dos trabalhos de pesquisa acabaram de ser publicadas em um novo artigo de  pesquisa (https://www.mdpi.com/2073-4409/13/1/31#). E o resultado é ainda mais emocionante, pois os pesquisadores utilizaram com sucesso a Edição Prime, uma forma de edição genética, que foi utilizada para corrigir uma mutação no gene RYR1 nas células musculares esqueléticas. Esta pesquisa fornece "prova de conceito" para a edição de genes como sendo uma estratégia em potencial para tratar miopatias relacionadas com RYR-1, que atualmente carecem de terapias eficazes. Segundo o cientista, "estes resultados são as primeiras demonstrações de que é possível corrigir mutações no gene RYR-1"
pesquisa (https://www.mdpi.com/2073-4409/13/1/31#). E o resultado é ainda mais emocionante, pois os pesquisadores utilizaram com sucesso a Edição Prime, uma forma de edição genética, que foi utilizada para corrigir uma mutação no gene RYR1 nas células musculares esqueléticas. Esta pesquisa fornece "prova de conceito" para a edição de genes como sendo uma estratégia em potencial para tratar miopatias relacionadas com RYR-1, que atualmente carecem de terapias eficazes. Segundo o cientista, "estes resultados são as primeiras demonstrações de que é possível corrigir mutações no gene RYR-1"
O gene RYR1 codifica um canal de cálcio denominado receptor 1 de Ryanodina, apresentada nas fibras musculares esqueléticas. A falha desse canal causa fraqueza muscular, que degenera acarretando deficiências motoras no indivíduo afetado. Atualmente, não existem tratamentos eficazes para estas miopatias, também conhecidas como doenças relacionadas ao RYR1, que são causadas principalmente por mutações pontuais. A Edição Prime permite a modificação precisa de nucleotídeos no DNA. Os resultados dos trabalhos de pesquisa pelos cientistas Kelly Godbout, Joël Rousseau e Jacques P. Tremblay, demostraram uma taxa de correção de 59% da mutação T4709M no gene RYR1 em mioblastos humanos pela entrega de RNA dos componentes de Edição Prime. Deve-se notar que o T4709M é recessivo e, portanto, as pessoas com mutação heterozigótica são saudáveis. Estes resultados são a primeira demonstração de que é possível corrigir mutações no gene RYR1.
A tecnologia de Edição Prime pode ser usada para corrigir mutações que causam miopatias relacionadas ao RYR1. Este grupo de doenças inclui a Hipertermia Maligna (HM), Miopatia Central Core (CCD), Miopaty Multi-Minicore (MmD), Miopatia Centronuclear (CNM), Desproporção Congênita do Tipo de Fibra (CFTD) e Rabdomiólise por Esforço (ERM). Até o momento, mais de 700 variantes no gene RYR1 foram identificadas. Este gene que codifica uma proteína chamada "receptor de rianodina 1" (RyR1), é o principal canal de cálcio no retículo sarcoplasmático (SR) nas fibras musculares esqueléticas. A disfunção desta proteína afeta o fluxo de cálcio para os músculos. A posição da mutação não afetará ou impactará na proteína, mas as mutações nos genes farão com que ocorra principalmente a um vazamento de cálcio. E como o cálcio é fundamental para a contração muscular, essa desregulação do RYR1 leva à fraqueza muscular, caibras, exaustão, intolerância ao calor, dificuldades respiratórias e até mesmo à reação maligna de hipertermia, ou Hipertermia Maligna. Essas miopatias, portanto, afetam gravemente a qualidade de vida dos pacientes. A proteína RYR1 tem variações funcionais limitadas, e o gene RYR1 é um dos mais intolerantes a variações de sequência no genoma humano.
Até o momento, não existe tratamento eficaz para essas doenças relacionadas ao RYR1. Como muitas mutações nos genes RYR1 são mutações pontuais, os resultados descritos no referido artigo demonstram claramente que a Edição Prime pode ser utilizada para corrigi-las, uma vez que pode substituir qualquer nucleotídeo do genoma.
O referido artigo relata a correção de uma dessas mutações (isto é, a T4709M) como exemplo. Esta mutação específica foi selecionada porque existe um modelo de camundongo (RYR1TM/Indel) com essa mutação que desenvolve sintomas claros. Confira o artigo científico no link -> https://www.mdpi.com/2073-4409/13/1/31#
TERAPIA GENÉTICA
A terapia genética é uma grande promessa para o tratamento de doenças genéticas, uma vez que aborda diretamente a raiz do problema. Ao corrigir mutações, a terapia genética tem o potencial de curar milhares de doenças hereditárias.
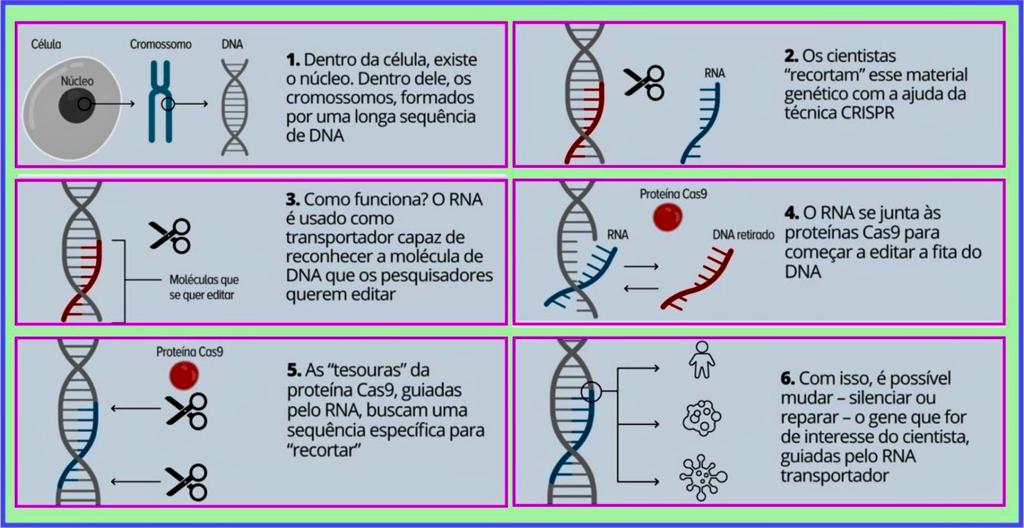
A descoberta do CRISPR/Cas9 em 2012 foi um marco no desenvolvimento de terapias genéticas. O Crispr/Cas9 é uma espécie de "tesoura genética", que permite à ciência mudar parte do código genético de uma célula. Com essa "tesoura", é possível, por exemplo, "cortar" uma parte específica do DNA, fazendo com que a célula produza ou não determinadas proteínas.
Este sistema usa uma nuclease Cas9 que induz uma quebra da fita dupla do DNA em um local preciso do genoma. Cas9 é direcionado para a sequência do genoma desejada por um único RNA guia (sgRNA). Este sgRNA é um RNA de fita simples complementar a uma sequência de DNA. A proteína Cas9 forma um complexo com o sgRNA e se liga a um motivo adjacente no DNA, induzindo um corte. Após a quebra da cadeia dupla no local do desejado, a célula irá reparar este corte por Reparação Dirigida por Homologia (HDR) se for fornecida uma sequência doadora. No entanto, a percentagem de correção de uma mutação precisa de nucleótidos por HDR é demasiado baixa para ser utilizada no tratamento de doenças hereditárias in vivo . Se nenhuma sequência doadora for fornecida, a célula reparará o corte por junção final não homóloga (NHEJ) e produzirá indels. InDels (inserções e deleções) são adições ou perdas de uma ou mais bases consecutivas na sequência do DNA.
CRISPR/Cas9
Em outubro de 2019, o grupo de David R. Liu publicou uma técnica notável chamada PRIME EDITION. Este sistema pode realizar inserções, deleções direcionadas e todas as 12 conversões de base possíveis.
O Prime Edition ou sistema Edição Prime (em português), é um método de edição de genoma que grava diretamente novas informações genéticas em um local (endereço) de DNA especificado usando uma endonuclease Cas9 prejudicada cataliticamente e fundida com uma transcriptase reversa projetada, programada com um RNA de guia Prime Edition (pegRNA) que especifica o local de destino e codifica a edição desejada. Esta tecnologia realiza modificações no DNA com precisão sem precedentes e oferece vantagens substanciais sobre o sistema tradicional CRISPR/Cas9.
Prime Editing é mais complexo que a edição CRISPR. Ele pode excluir comprimentos longos de DNA causador de doença ou inserir DNA para reparar mutações perigosas, tudo sem desencadear as respostas caóticas (e possivelmente prejudiciais) do genoma introduzidas por outras formas de CRISPR.
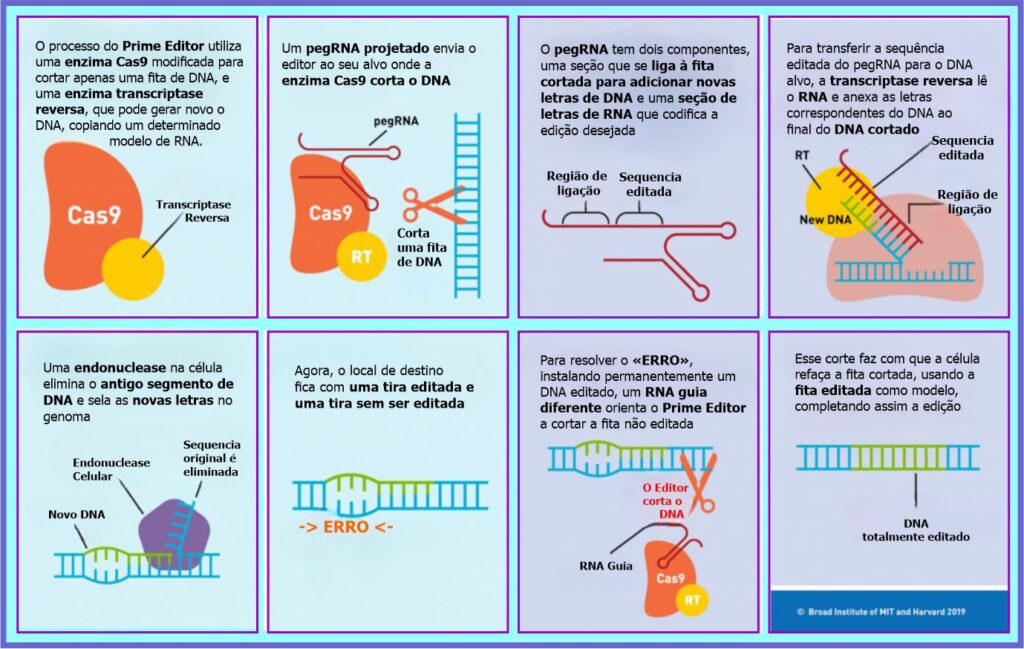
Prime Edition
Em resumo, a técnica CRISPR-Cas9, popularmente utilizada para modificação genética pela comunidade científica, baseia-se na atividade nuclease da enzima Cas9 que corta as duas fitas de DNA, e utiliza a maquinaria de reparo de danos da própria célula. No entanto, o sistema de reparo pode inserir ou deletar letras de DNA, causando efeitos inesperados. Já a nova tecnologia “Prime Editing” ou Edição Prime utiliza uma versão enzima Cas9 que além de reconhecer sequências específicas de DNA, corta apenas uma das fitas da dupla-hélice. Dessa forma, a edição ocorre no local correto do corte através da ação de uma enzima transcriptase e uma fita de RNA guia (pegRNA).

