Na última postagem entitulada, A Importância das Intervenções No Estilo de Vida Para Controle Dos Sintomas da Miopatia Congênita Centronuclear, fiz uma abordagem sobre a importância das condutas multidisciplinar e individualizada, focada na reabilitação e no suporte contínuo para otimizar a funcionalidade e o bem-estar. Esse estilo de vida, seria sustentado por um tripé de condutas, sintetizado no Controle de Stress, Nutrição, e Atividade Física.
Seguindo essa linha de entendimento, gostaria de compartilhar com o leitor sobre minha experiência pessoal, enquanto portador de Miopatia Congênita Centronuclear causada pela mutação genética no RYR-1 com herança autossômica recessiva, com o acompanhamento nutricional que faço com a Dra Victoria Pitombo. Como tanto, segue as gentis palavras da Dra Victoria sobre nossa relação enquanto paciente, e logo a seguir você verá um estudo, base do trabalho aplicado no meu programa nutricional e de suplementação.
" Desde o início do nosso acompanhamento, o que mais me marca no Orlando é o comprometimento real que ele tem com a própria saúde. A nossa relação vai muito além de prescrições: é uma parceria construída com confiança, escuta e respeito por um corpo que exige atenção minuciosa devido à Miopatia Congênita Centronuclear. Cada ajuste que eu faço, desde a ingestão proteica até orientações de digestão e hidratação, tem um propósito maior: proteger sua musculatura, preservar funcionalidade, reduzir estresse oxidativo e garantir mais autonomia e qualidade de vida para ele.
Ao longo desses meses, construímos uma nutrição precisa e totalmente personalizada: calorias calibradas para preservar massa muscular e reduzir gordura abdominal, além de suplementos escolhidos para otimizar função neuromuscular sem gerar desconforto gastrointestinal. Tudo específico para o Orlando. Ver a evolução dele, melhora da autoestima, engajamento e confiança no processo (que ainda temos muito pela frente!) é uma das partes mais gratificantes do nosso trabalho juntos. " Dra Victoria Pitombo
NOTA IMPORTANTE: O estudo apresentado a seguir deve ser entendido como elemento de base científica, portanto, o programa de nutrição e suplementação para cada indivíduo deve ser altamente individualizado e baseado na avaliação clínica, estado nutricional atual e exames laboratoriais. Para garantir segurança e eficácia, as intervenções nutricionais e a suplementação devem ser sempre orientadas e conduzidas por um médico e um nutricionista especializados ou com conhecimento em doenças neuromusculares.
Dra. Victoria C. Pitombo - CRN 58214
℘ Nutricionista especializada a nível de Residência em doenças crônicas pelo Hospital do Coração (Hcor)
℘ Nutricionista Funciona(VP), Esportiva e com especialidade em Obesidade (USP)
Nutrição e Suplementação na Miopatia Congênita Centronuclear Associada à Mutação no Gene RYR1: Evidências Atuais e Considerações Clínicas
As miopatias relacionadas ao gene RYR1 englobam o grupo mais prevalente de miopatias congênitas, incluindo fenótipos como miopatia centronuclear. A mutação compromete a função do receptor rianodina, responsável pela liberação controlada de cálcio intracelular durante a contração do músculo. Como resultado, observam-se fraqueza de predomínio proximal, fadiga rápida, intolerância ao exercício, risco maior de hipertermia maligna e, em muitos casos, comprometimento respiratório.
Embora ainda não exista uma cura, percebe-se consenso crescente de que intervenções nutricionais e suplementação direcionada desempenham um papel importante no tratamento das miopatias. Isso inclui uma melhora na preservação da função muscular, na proteção óssea e na modulação do estresse oxidativo, todos aspectos centrais na fisiopatologia dessa condição.
1. Fundamentos do manejo nutricional
Adequação energética e composição corporal
Pacientes com miopatias congênitas apresentam gasto energético variável, podendo evoluir com desnutrição ou excesso de peso. Ambos são prejudiciais à função muscular e respiratória. Assim, a ingestão energética deve ser individualizada, sempre com foco na preservação da massa magra.
Proteína e estímulo anabólico
A literatura mostra que em doenças neuromusculares a ingestão proteica deve ser 1,2–1,5 g/kg/dia, distribuída ao longo do dia. Para alguns pacientes, pode-se extrapolar a ingestão para até 2g/kg/dia.
A distribuição da proteína ao longo do dia (em no mínimo 4 refeições), otimiza a síntese proteica muscular e reduz períodos de catabolismo, especialmente importante em indivíduos com fadiga crônica e menor resposta anabólica.
2. Suplementação baseada em plausibilidade fisiológica e evidência
Creatina Monohidratada
A creatina possui o potencial de aumentar a disponibilidade de fosfocreatina e melhora a ressíntese de ATP, mecanismo útil em condições que cursam com fadiga muscular rápida. Ensaios clínicos em outras doenças neuromusculares demonstram melhora modesta, mas consistente, na força e na resistência.
Dose usual: Em pacientes com Miopatia congênita centronuclear, o ideal é calcular a dose por kg de peso. Exemplo: Se o paciente pesa 67kg ele deve consumir 67kg x0,1 = 6.7g de creatina por dia.
Leucina
A leucina ativa a via mTOR, estimulando a síntese proteica muscular que é um processo frequentemente diminuído em miopatias com limitação funcional.
Na maioria das vezes, só o suporte de leucina via alimentos não é suficiente, então é interessante suplementar. Dose utilizada em estudos: 2–3 g junto com as refeições principais 2 a 3x por dia, dependendo do paciente. Ou, o paciente pode consumir a leucina 1x ao dia no período noturno.
N-acetilcisteína (NAC)
O estresse oxidativo é um dos mecanismos envolvidos na disfunção do RYR1. A NAC é precursora de glutationa e moduladora do sistema redox. Ensaios clínicos mostram segurança, embora os efeitos em marcadores oxidativos sejam variáveis.
Dose usual: 600–1200 mg/dia.
Outros antioxidantes relevantes
O desequilíbrio redox presente em mutações do RYR1 motiva o uso de antioxidantes adjuvantes:
Vitamina B12
A deficiência de B12 agrava fadiga, fraqueza e neuropatia, podendo intensificar limitações motoras já presentes.
Indicação: quando há deficiência documentada ou fatores de risco.
Doses usuais: 500–1000 mcg/dia (oral) ou protocolo IM médico.
Vitamina D e Cálcio - papel central para músculo e osso
Vitamina D
Há maior prevalência de deficiência de vitamina D em doenças neuromusculares, associada a redução da força, pior função respiratória e menor densidade óssea.
A suplementação e doses deve ser guiada por dosagem sérica (25-OH).
Cálcio
Por que o cálcio é tão importante nesses pacientes ?
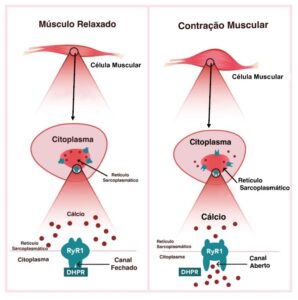
O cálcio é o íon mais importante da contração muscular e justamente o elemento cuja movimentação é regulada através do receptor rianodina (RYR1). Em pacientes com mutações nesse gene, ocorrem algumas alterações no fluxo de Ca²⁺, como liberação excessiva ou dificuldade de recaptura, contribuindo para: fadiga precoce;maior risco de dano estrutural das fibras; dificuldade de relaxamento muscular e piora de sintomas ao esforço.
Além disso, muitos pacientes apresentam baixa densidade mineral óssea devido à imobilidade, baixa massa muscular, ingestão reduzida ou pouca exposição solar. Assim, o cálcio torna-se essencial tanto para a função muscular quanto para a prevenção de osteopenia e fraturas, que impactam diretamente mobilidade e autonomia.
Recomendações gerais:
L-Tirosina
A L-tirosina é precursora de catecolaminas e pode modular aspectos de vigília, foco e tolerância ao esforço, elementos frequentemente prejudicados em miopatias crônicas. Estudos sugerem benefício potencial em quadros de fadiga central.
Dose usada em protocolos clínicos: 500–2000 mg/dia.
Considerações finais
Embora a miopatia centronuclear por mutação no RYR1 não possua ainda um tratamento curativo, o manejo nutricional adequado e a suplementação estratégica apresentam papel importante no suporte à função muscular, no controle do estresse oxidativo, na proteção óssea e na melhoria da capacidade funcional diária.
Intervenções como adequação proteica, creatina, leucina, vitamina D e cálcio, NAC, antioxidantes, vitamina B12 e L-tirosina possuem fundamentos fisiológicos consistentes e vêm sendo incorporadas progressivamente à prática clínica, sempre de forma individualizada e integrada ao acompanhamento multiprofissional.
O estilo de vida levado por um indivíduo afetado por uma miopatia congênita é fundamental para o controle dos sintomas da condição, embora não existam tratamentos específicos para a cura da doença. Essas intervenções são consideradas tratamentos de suporte e visam preservar a função muscular, maximizar a independência, e melhorar a qualidade de vida do paciente.
O tratamento da miopatia congênita deve ter uma abordagem multidisciplinar e individualizada, focada na reabilitação e no suporte contínuo para otimizar a funcionalidade e o bem-estar. Esse estilo de vida a que me refiro, seria sustentado por um tripé de condutas, sintetizado no Controle de Stress, Nutrição, e Atividade Física.
 Controle do Stress
Controle do Stress
O controle do stress (stress management) é de extrema importância para a manutenção da saúde e qualidade de vida de indivíduos portadores de uma miopatia congênita. Embora não haja a cura ou tratamento específico para a doença em si, muitas vezes a única terapia indicada é de suporte, como fisioterapia.
Contudo, existem outras condutas que podem ser tomadas visando a mitigação dos sintomas da doença, melhoria na qualidade de vida dos indivíduos, e que inclusive interferem positivamente no prognóstico da doença. Essa conduta diz respeito a uma série de mudança ou incorporação de hábitos vida, chamada de Controle de Stress, e que é composta em vários fatores interligados:
 Nutrição
Nutrição
A qualidade de vida de um indivíduo afetado por uma miopatia é fortemente influenciada pela capacidade de manter a funcionalidade e a autonomia o máximo possível, apesar da fraqueza muscular, que é o sintoma mais comum. Neste contexto, a importância do controle nutricional é crítica e multifatorial, sendo uma parte essencial para seu tratamento de suporte.
No caso específico da Miopatia Centronuclear, por ser uma doença neuromuscular que causa fraqueza muscular significativa (hipotonia), pode levar a várias complicações que exigem um acompanhamento nutricional individualizado e rigoroso, fundamental para minimizar a perda de massa muscular, garantir o aporte energético adequado, e prevenir complicações secundárias à fraqueza. Esse manejo nutricional não visa curar a doença, mas sim otimizar a saúde geral, apoiar a função muscular e respiratória, e prevenir complicações que impactam severamente a qualidade de vida.
Não existe uma dieta ou protocolo de suplementação único para todos os indivíduos com miopatia congênita. A dieta e suplementação deve garantir um aporte energético suficiente para as necessidades metabólicas e para preservar a já tão sofrida massa muscular. O cálculo do gasto energético total precisa ser individualizado, considerando o nível de atividade física e a gravidade da miopatia.
O plano deve ser altamente individualizado e baseado na avaliação clínica, estado nutricional atual e exames laboratoriais. Para garantir segurança e eficácia, as intervenções nutricionais e a suplementação devem ser sempre orientadas por um médico e um nutricionista especializados ou com conhecimento em doenças neuromusculares. Esse planejamento nutricional se baseia em algumas premissas básicas:
Em resumo, uma nutrição bem planejada e a suplementação direcionada são ferramentas de suporte poderosas que visam otimizar a função muscular residual, prevenir deficiências nutricionais, apoiar a saúde óssea e melhorar a qualidade de vida do indivíduo com miopatia congênita.
 Atividade Física
Atividade Física
Sempre digo que a atividade física, no contexto das miopatias congênitas, vai além da função de mero suporte clínico, pois ela se estabelece como um elemento fundamental na promoção da qualidade de vida. A atividade física é crucial não apenas para retardar o declínio funcional, mas também para promover o bem-estar psicossocial do indivíduo afetado pela doença.
Para indivíduos com miopatia congênita, a atividade física é uma ferramenta terapêutica que busca promover a capacidade do indivíduo agir de forma mais autônoma possível, além de atuar na conexão social e dignidade pessoal, situações estas tão importantes quanto qualquer intervenção médica para alcançar uma qualidade de vida.
A atividade física capacita o indivíduo, dando-lhe um sentimento de propósito, controle e autoestima, que são um pilares da qualidade de vida.
A Declaração de Consenso sobre Padrão de Cuidados para Miopatias Congênitas, publicada em 2012 no Journal of Child Neurology, tratou a atividade física no contexto de se enfatizar a importância de um programa estruturado, mas altamente individualizado, para manter a função e a mobilidade.
O consenso reconheceu que, embora não exista cura, a fisioterapia e a terapia ocupacional (TO) são cruciais para o manejo da doença, e como promoção da qualidade de vida do indivíduo.
As diretrizes do consenso de 2012 sobre atividade física e reabilitação se referiam principalmente aos seguintes pontos:
Em essência, a declaração de consenso enfatizou que a atividade física para miopatias congênitas não é uma atividade de "ganho de força" típica, mas sim uma intervenção de manutenção e reabilitação focada em preservar a função e evitar as complicações secundárias da fraqueza, mas sobretudo e principalmente como agente de promoção de qualidade de vida.
Desde que nasci, eu fui desafiado pelos sintomas de uma doença que me fazia ser diferente de outras crianças, ela me confrontava a todo tempo, me causava limitações físicas funcionais progressivas, e assim, causava a mim e à minha família um turbilhão de questionamentos e dúvidas sobre do que se tratava. Diante dos diagnósticos e prognósticos que recebia, dos mais piores possíveis, assim como pela falta de uma solução com um possível tratamento, eu, com toda minha inocência e intuito, resolvi que a melhor coisa que tinha a fazer era viver, e viver com intensidade, buscando a superação das minhas limitações, potencializando assim minha capacidade com o que tinha de físico, emocional, e intelectual. E sempre digo que apesar de tudo, eu nunca me vi no espelho como uma pessoa diferente, e neste contexto de vida, brinquei, namorei, me formei, casei, trabalhei muito, tive dois filhos, enfim, enquanto isso o mundo foi girando, a ciência descobrindo e explicando as coisas, até que quando tinha 44 anos, obtive meu diagnóstico sobre minha doença, ou seja, que tinha uma mutação no gene RYR1, e que me causava uma doença chamada de Miopatia Congênita Centronuclear, e que isso é que era a responsável pela condição que vivia desde que nasci. Pois bem, não mudou nada na minha vida, a não ser que descobri o endereço e nome da minha doença, o resto foi que ela era uma "Doença Rara", incurável e sem tratamento, e por fim, que pouco se conhecia sobre ela no meio médico-científico.
Sempre fui uma pessoa muito resiliente, que não só tem a força interior para enfrentar a realidade, por mais dura que seja, mas também tem a ambição intelectual e pessoal para moldar ativamente minha realidade em uma vida melhor, ou como diz minha mãe, mais "suave". Sempre fui uma pessoa da mente muito aberta ao conhecimento, ao questionamento, à observação, ao aprendizado, e essas minhas características não me fizeram ser uma pessoa acomodada. Não saber a princípio qual era doença que me acometia, e posteriormente descobri-la, e o pior que não tinha cura ou tratamento, nunca me causou sentimentos negativos, muito pelo contrário, me impulsionou a buscar viver melhor, mas também ao conhecimento.
essas minhas características não me fizeram ser uma pessoa acomodada. Não saber a princípio qual era doença que me acometia, e posteriormente descobri-la, e o pior que não tinha cura ou tratamento, nunca me causou sentimentos negativos, muito pelo contrário, me impulsionou a buscar viver melhor, mas também ao conhecimento.
A busca pelo conhecimento teórico sobre o mecanismo de funcionamento do gene, o RYR1, e como ele interferia no meu corpo foi um grande marco para meu conhecimento intelectual, assim como satisfação interna para minhas ansiedades. Contudo, o conhecer de perto, praticamente quase podendo o “tocar”, foi uma de minhas maiores experiências que já tive nesta minha peregrinação em busca de saber sobre a doença que me acomete desde que nasci. E isso se deu, depois de muito tempo, especificamente, recentemente, na última Conferência de Família e Workshop de Pesquisa do RYR1, realizada em julho de 2025 em Pittsburgh, EUA, durante uma reunião particular com o Dr. Filip Van Petegem. Na oportunidade, pude assistir um vídeo com a reconstituição em 3D “do meu RYR1”, ao mesmo tempo que o escutava fazer uma explanação específica sobre minha mutação. Dr. Van Petegem lidera um laboratório de pesquisa no Departamento de Bioquímica e Biologia Molecular na Universidade da Colúmbia Britânica (UBC), Vancouver, Canadá, no qual utiliza a cristalografia de raios X e microscopia crioeletrônica para estudar suas estruturas 3D, o que proporciona estudar a estrutura e função dos canais iônicos, com foco no músculo cardíaco e esquelético, e isso inclui o Receptor de Ryanodine (RyR). Um dos trabalhos do Dr Petegem consiste na importante abordagem em determinar as estruturas tridimensionais muito detalhadas desses canais do RYR1, permitindo a análise dos efeitos diretos das mutações na estrutura, e consequente interferência física no indivíduo afetado.
A título ilustrativo e como exemplo, gostaria de compartilhar com o leitor dessa postagem um pequeno vídeo criado pelo Dr Van Petegem, a partir da técnica acima descrita, da variante do meu RYR1 com a respectiva mutação genética. O vídeo lhe permitirá entender melhor a complexidade da estrutura molecular do gene RYR1, descrita na postagem anterior, O GIGANTE DO GENOMA QUE FAZ OS MÚSCULOS SE MOVEREM, e que neste caso se trata do meu RYR1.
O Vídeo
Para orientar um pouco melhor a apresentação deste vídeo, e assim entender o que acontece com meu gene RYR1, veja a seguir algumas da partes importantes da apresentação:
1). cada esfera que você vê representa um átomo individual;
2). o vídeo começa com uma visão 'top', ou seja, por cima do RYR1. Olhando no fundo da parte central, está a região chamada de 'poro', onde os íons de cálcio podem passar;
3). o vídeo amplia a localização da minha variante, E1175K. O aminoácido do 'tipo selvagem' é o chamado 'E', também conhecido como 'Glu' ou 'Glutamato'. Este aminoácido tem a propriedade especial de carregar carga negativa. No vídeo, este E1175 é mostrado em vermelho;
4). pode-se ver que o E1175 está apontando para outro aminoácido em uma cor diferente, em azul. Esse aminoácido, conhecido como 'R' ou 'Arg' ou 'Arginina', é especial porque carrega uma carga positiva. Assim, em pessoas sem nenhuma variante RYR1, haverá normalmente uma atração entre o 'E' carregado negativamente em vermelho, e o 'R' carregado positivamente em azul;
5). na variante demonstrada (a minha no caso), o E1175 carregado negativamente é substituído por 'K'. Este aminoácido, também conhecido como 'Lys' ou 'Lysine', também carrega carga positiva. Portanto, a variante não apenas abole a interação positivo-negativa normal, mas também coloca um aminoácido carregado positivamente (K) ao lado de outro (R), e isso desestabiliza a proteína. Por causa disso, o RYR1 se torna mais móvel e o poro pode abrir mais facilmente, ou seja, não funcionar da maneira correta. Esse defeito leva a um 'vazamento' de cálcio, que pode danificar o funcionamento normal dos músculos.
(VIDEO 1)
(VIDEO 2)
As doenças relacionadas ao RYR1 são identificadas com base em sua classificação histopatológica, isto é, pela aparência da biópsia do músculo na lâmina do microscópio. A diferenciação encontrada na biópsia designará o tipo da doença, se é por exemplo, Miopatia Central Core, Miopatia Multiminicore, Miopatia Centronuclear, ou Desproporção Congênita de Tipos de Fibras.
A Miopatia Congênita Centronuclear (MCCN) e a Miopatia Central Core (MCC) são ambas doenças musculares hereditárias causadas por mutações genéticas. A MCCN pode estar associada a mutações em diferentes genes, no DNM2, BIN1, MTM1, e RYR1, já a MCC está associada somente ao gene RYR1.
Esses tipos de doenças relacionadas ao RYR1 variam amplamente em termos dos seus diferentes sinais e sintomas, de quando eles inicialmente se apresentaram, além da sua respectiva gravidade. Embora sejam altamente variáveis, os sintomas presentes também dependem se a mutação do gene RYR1 é autossômica dominante ou autossômica recessiva.
Uma pergunta que sempre chega até mim é sobre as diferenças entre a Miopatia Centronuclear e a Miopatia Central Core. Assim, eu, enquanto portador da Miopatia Congênita Centronuclear (MCN), buscarei esclarecer pontualmente neste texto, me atendo ao tipo que me acomete, que é a pela mutação no gene RYR1.
Estas doenças apesar de terem sintomas parecidos e compartilharem de algumas características clínicas em comum, se confundem entre si, e apresentam com algumas diferenças distintas:
Miopatia Congênita Centronuclear (MCCN)
Miopatia Central Core (MCC)
Em resumo, a Miopatia Congênita Centronuclear e a Miopatia Central Core, ambas doenças relacionada ao RYR1, têm seus sintomas e sinais físicos que podem se parecer, podem se confundir, mas são diferentes, a contar da análise histológica das células musculares em uma biópsia, exame este que é crucial para diferenciar entre as duas condições e determinar o diagnóstico correto, conduta médica, tratamento, e até prognóstico de evolução.
Nota: todo o material escrito nesta página é oriundo de pesquisa científica, e conclusões próprias do autor deste website, que convive como portador, desde o nascimento, com a Miopatia Congênita Centronuclear causada pela mutação no gene RYR-1
As miopatias são doenças que afetam os músculos, e podem se apresentar desde o nascimento até a idade adulta. Quando a miopatia se manifesta no início da vida, é frequentemente referida como miopatia congênita e de origem genética.
O diagnóstico de miopatia em uma criança geralmente envolve uma combinação de exame físico, história clínica, testes laboratoriais, incluindo biópsia e exame genético, além de exames de imagem. O diagnóstico preciso depende da avaliação completa feita por um médico especialista em doenças neuromusculares pediátricas.
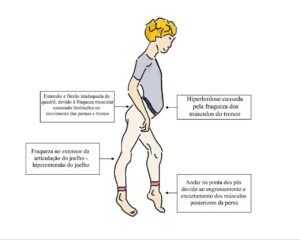
Existem alguns sinais físicos que são característicos em uma criança portadora de miopatia. Além do Movimento de Gowers, já descrito nesta website, o Sinal de Trendelenburg e a Marcha Miopática também são sinais físicos que podem ser cruciais no exame clínico quando do diagnóstico de uma miopatia. Esses sinais são incorporados como característica da doença, podendo ter um impacto significativo na qualidade de vida de quem a sofre, portanto, o acompanhamento e tratamento adequados são essenciais para ajudar os indivíduos a controlar os sintomas, limitações, evitar complicações secundárias, e manter uma boa qualidade de vida.
A Marcha Miopática é um termo médico usado para descrever um padrão de caminhada que é afetado pela fraqueza muscular causada por uma miopatia, que também é conhecida como Marcha do Pato. Essa condição provoca uma série de alterações na maneira de andar dos indivíduos afetados. Um dos sintomas mais característicos é também chamado de marcha bamboleante. Esta marcha é caracterizada pelo balanço do tronco e separação dos pés durante a caminhada. Como os músculos do tronco também são afetados, desencadeia-se uma postura anormal, apresentando como exemplo a hiperlordose lombar, que é uma curvatura excessiva na região lombar. Outra característica distintiva é andar na ponta dos pés, que provoca o espessamento e retração da musculatura posterior da perna, criando uma aparência de pseudo-hipertrofia. Nesta condição, os músculos tornam-se mais espessos e volumosos, mas na verdade é resultado da destruição da fibra muscular e da sua substituição por tecido fibroso ou fibras colágenas. Este padrão de marcha é uma característica comum em várias formas de miopatia e pode variar em gravidade dependendo do tipo e da progressão da doença.
O Sinal de Trendelenburg é uma característica bem comum em indivíduos afetados por uma miopatia, com fraqueza da musculatura abdutora do quadril, em especial o glúteo médio. O nome deste sinal é em homenagem ao cirurgião alemão Friedrich Trendelenburg. O sinal de Trendelenburg é positivo se, quando o quadril de um paciente que está de pé sustentado por somente uma perna, cai para o lado da perna levantada. A fraqueza é presente no lado da perna em contato com o chão. O corpo não é capaz de manter a perna estável, causando uma inclinação ou queda da pelve em comparação com o lado contralateral. Essencialmente, o Sinal de Trendelenburg é causado pela fraqueza dos músculos glúteo médio e mínimo.
O Sinal de Trendelenburg e a Marcha Miopática, apesar de poderem variar dependendo da miopatia, e da forma que ela se apresenta em cada indivíduo, ao longo do tempo, podem desencadear uma série de consequências no corpo (ufa ! …e eu que o diga…), podendo relacionar algumas delas:
Nota: todo o material escrito nesta página é oriundo de pesquisa científica, e conclusões próprias do autor deste website, que convive como portador, desde o nascimento, com a Miopatia Congênita Centronuclear causada pela mutação no gene RYR-1
A Miopatia Congênita Centronuclear (MCCN) é uma doença muscular congênita rara caracterizada por fibras celulares com núcleos centralizados proeminentes em biópsias musculares. A doença é clinicamente heterogênea, variando de fenótipos hipotônicos graves já no nascimento até fraqueza muscular leve com início na idade adulta, e pode ter múltiplos modos de herança em associação de causa por mutações nos genes MTM1, DNM2, BIN1 e RYR1.
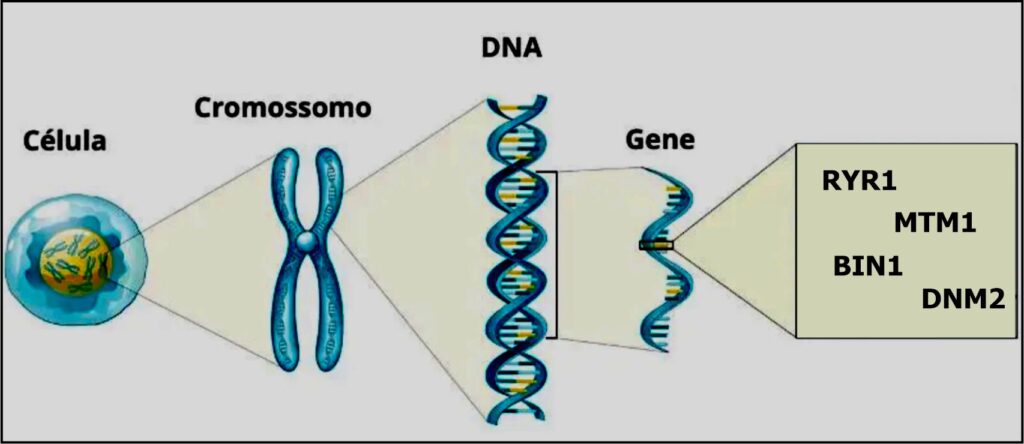
Assim como existe uma grande complexidade no diagnóstico de uma miopatia, tema esse abordado em outra postagem no SORRYR-1.com.br, essas diferentes causas, são também motivo de grande confusão em diagnósticos. É importante consultar um médico especialista para um diagnóstico preciso e assim obter informações detalhadas sobre a mutação específica no caso individual. As diferentes mutações genéticas que causam a Miopatia Congênita Centronuclear podem resultar em variações na gravidade, evolução e sintomas da doença. É importante dizer que a gravidade dos sintomas da doença pode variar de pessoa para pessoa, mesmo com a mesma mutação genética.
Seguem as diferentes causas de origem da Miopatia Congênita Centronuclear (MCCN):
O gene MTM1 é responsável pela codificação de proteína chamada miotubularina. Essa proteína desempenha um papel importante na função muscular, fundamental por atuar como uma enzima fosfatase de desempenho crítico na regulação do tráfego de vesículas dentro das células musculares, particularmente nas fibras musculares esqueléticas. Quando há uma mutação no gene MTM1, a produção ou função da miotubularina é afetada, e sso pode resultar em um acúmulo anormal de vesículas dentro das fibras musculares, levando à fraqueza muscular e outros sintomas associados à Miopatia Congênita Centronuclear.
A dinamina 2, codificada pelo gene DNM2, desempenha um papel crucial na regulação do tráfego de vesículas que transportam proteínas essenciais para a função muscular normal. Ela ajuda a controlar a fusão e divisão dessas vesículas, permitindo a entrega adequada de proteínas contráteis, como a miosina e actina, aos locais onde são necessárias para a contração muscular.
Quando ocorrem mutações no gene DNM2, a função da dinamina 2 pode ser comprometida, resultando em distúrbios que pode levar a fraqueza muscular, em especial nos músculos proximais, e outros sintomas associados a condições da MCCN. Portanto, a dinamina 2 desempenha um papel importante na manutenção da função muscular saudável.
O gene BIN1 codifica a proteína anexina A2, que está envolvida na regulação das membranas celulares e no tráfego de vesículas nas células musculares. Essas funções desempenhadas pela anexina A2 são essenciais para a saúde e a função das fibras musculares, a mutação nesse gene afeta negativamente a estrutura e a função das células musculares, o que resulta em fraqueza muscular e outros sintomas associados à CCNM. A gravidade e a apresentação dos sintomas podem variar com base na mutação específica do gene BIN1 envolvida.
O gene RYR1 codifica o receptor de rianodina 1, que é uma proteína essencial para a função das fibras musculares. O receptor de rianodina está envolvido na liberação (válvula de controle) de cálcio das reservas intracelulares, um processo fundamental para a contração e relaxamento muscular.
As doenças musculares são aquelas que afetam a estrutura e funcionamento do músculo, sendo as principais: as distrofias musculares, as miopatias congênitas, as miopatias inflamatórias e as miopatias endócrinas e metabólicas. É importante destacar que cada uma delas possui suas variações que também se diferenciam.
Essas doenças já foram muito confundidas em diagnósticos no passado, e fico triste, porque isso ainda tem acontecido nos dias de hoje, mesmo com os avanços científicos. A única razão que acredito ser ainda a causa para essa confusão nesses diagnósticos seria por essas doenças serem consideradas “doenças raras”, portanto, muitas vezes desconhecidas por parte da comunidade médica. Assim, pode haver a falha no momento dos exames clínicos, ponto inicial para diagnóstico de qualquer doença.
Eu mesmo vivi uma experiência dessa, pois no decorrer de grande parte da minha vida eu recebi vários “diagnósticos” de Distrofia Muscular Congênita (DMC) do tipo: Duchenne, Facioescapuloumeral e Cinturas. E, os prognósticos foram do pior a até o mais brando. Estes diagnósticos ou hipóteses de diagnósticos vieram até de importantes instituições, como de uma clínica indicada pelo MDA (Muscular Distrophy Association), maior referência ligada a essa doença.
Deve-se levar em consideração que, naquela época, pouco se sabia sobre essa doença, nem tão pouco sobre a genética humana; contudo, um erro de diagnóstico hoje em dia seria inaceitável. Essa situação causou em mim grandes transtornos, de emocional aos físicos. Somente aos 44 anos de idade foi que finalmente obtive meu correto e “definitivo diagnóstico”, ou seja, de que sou portador de Miopatia Congênita Centronuclear (MCC), causada pela mutação no gene RYR1.
A Distrofia Muscular Congênita e a Miopatia Congênita Centronuclear apresentam várias características em comum, tais como: são doenças de origem genética, afetam os músculos esqueléticos, caracterizam-se clinicamente por hipotonia e fraqueza muscular, geralmente apresentam-se desde o nascimento, têm curso clínico estático ou lentamente progressivo. Essas doenças não tem cura, e o tratamento envolve terapia de suporte, como fisioterapia, dispositivos de mobilidade e, em alguns casos, medicamentos. Mesmo assim, as duas doenças neuromusculares diferem entre si.
Daí, eu volto com a questão sobre as falhas nos diagnósticos, já que muitos médicos se prendem somente ao resultado do exame genético e não conhecerem os sinais clínicos das diferentes doenças e particularidades dos indivíduos afetados.
Assim, essas noções devem ser levadas em consideração por três razões: Primeiro, muitas das miopatias congênitas podem ser causadas por mutações em mais de um gene, o que sugere um impacto da heterogeneidade genética. Segundo, mutações no mesmo gene podem causar diferentes patologias musculares. Terceiro, a mesma mutação genética pode levar a diferentes características patológicas e sintomatológicas em membros da mesma família ou no mesmo indivíduo em idades diferentes.
Em resumo, eu destacaria que tanto a Distrofia Muscular, quanto a Miopatia Congênita Centronuclear são de origem genética, mas distintas em termos de suas características clínicas e podem variar em gravidade de pessoa para pessoa. Enquanto a Distrofia Muscular envolve a degeneração progressiva dos músculos devido a problemas na estrutura das proteínas musculares, a Miopatia Congênita Centronuclear é caracterizada por uma anormalidade na localização dos núcleos das células musculares. Assim, é importante consultar um médico especialista para um diagnóstico preciso, para que se possa ser feito um acompanhamento adequado do caso, pois o tratamento pode variar dependendo da condição clínica específica de cada indivíduo.
Os portadores de doenças relacionadas ao RYR-1 através do SORRYR-1 comemoram junto com a comunidade dos portadores de Distrofia Muscular, em especial os Duchennes, pela recente aprovação pelo FDA (Federal Drug Administration) do ELEVIDYS (delandistrogene moxeparvovec-rokl) destinada ao tratamento de pacientes pediátricos ambulatorial com idades entre os 4 e os 5 anos com Distrofia Muscular tipo Ducehene #Duchenne #MuscularDystrophy (#DMD) com mutação confirmada no gene DMD.
O SORRYR-1 cumprimenta também a Sarepta Therapeutics @sareptatherapeutics pelo importante trabalho em pesquisas e desenvolvimento de medicamentos genéticos de precisão potencialmente transformadores para doenças raras. O ELEVIDYS é a primeira terapia gênica para DMD (Distrofia Muscular tipo Ducehene) e foi projetada para atingir a causa subjacente da doença.
O SORRYR-1 destaca a crucial importância do MDA (Muscular Dystrophy Association) @mdaorg que esteve presente desde o início nos trabalhos de pesquisas da terapia genética, e criou recentemente uma Rede de Apoio à Terapia Gênica (MDA Gene Therapy Support Network - GTx) para fornecer recursos e orientação para médicos e famílias. O MDA (Muscular Dystrophy Association) é a primeira organização de saúde voluntária nos Estados Unidos voltada para pessoas que vivem com distrofia muscular, e doenças neuromusculares, e que a mais de 70 anos abriu o caminho para acelerar as pesquisas, promoção do atendimento e defesa ao apoio de famílias e indivíduos afetados.
O SORRYR-1 entende esta grande conquista para o tratamento dos portadores de Duchene como uma grande prova dos avanços científicos e empenho da indústria farmacêutica, reforçando assim, a esperança de nós, portadores de doenças relacionadas à mutação no RYR-1, de que uma terapia semelhante possa surgir em um futuro próximo em nosso benefício como tratamento e cura.
TERAPIA GENÉTICA
A terapia genética trata uma doença agindo no próprio gene mutado. A terapia genética pode introduzir um gene para ajudar a combater doenças, substituir um gene mutante, editar um gene mutante ou excluir um gene mutante. Em teoria, as terapias genéticas oferecem o potencial de cura, não apenas de tratamento.
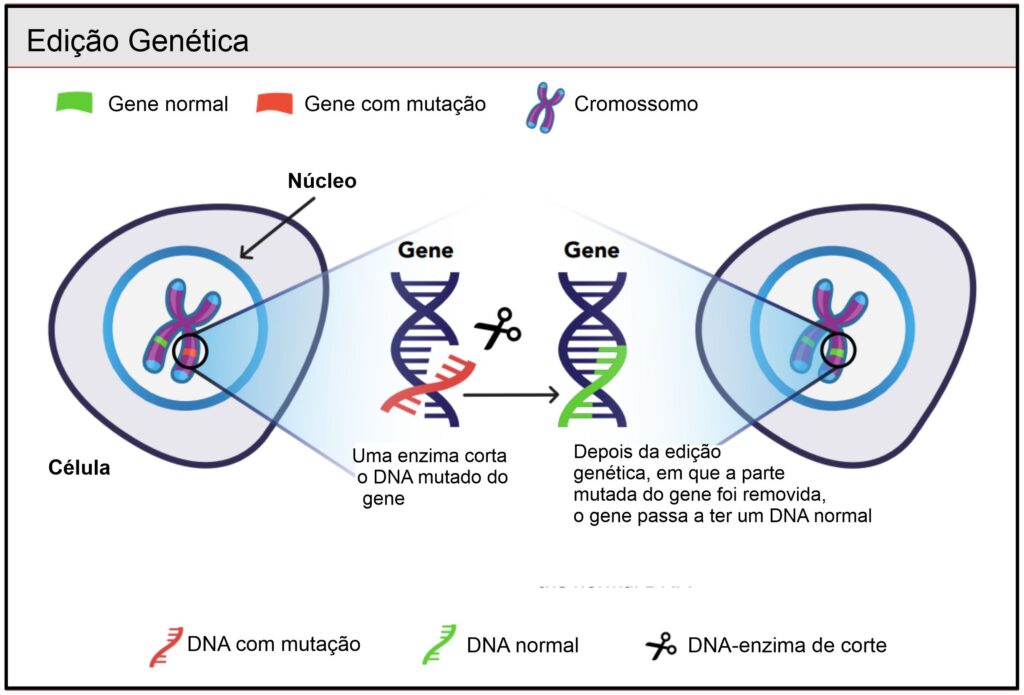
Edição de Gene
Na terapia genética, a edição de genes tem o objetivo de corrigir ou “editar” apenas uma pequena parte do gene, sendo esta, uma abordagem que visa tornar as mudanças mais precisas e permanentes.
RYR-1 Foundation Clinical Care Guidelines
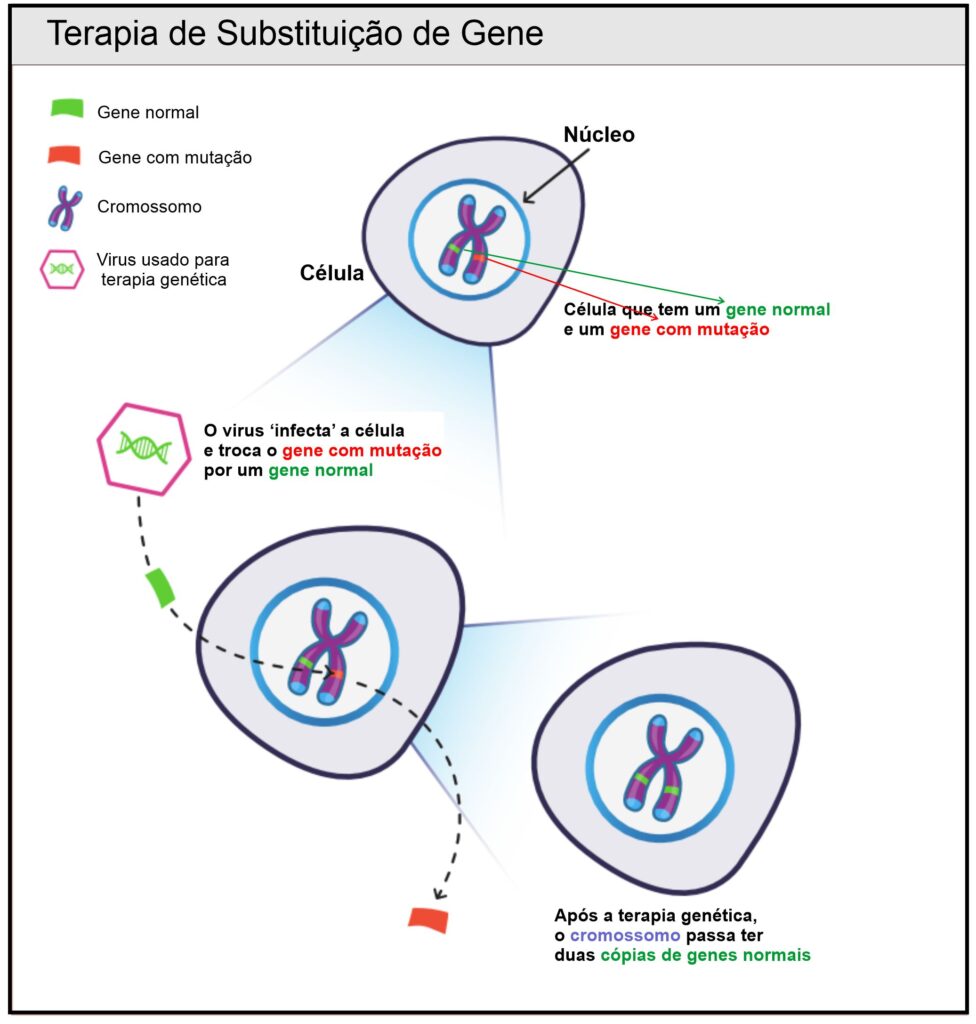
Terapia de Substituição de Gene
A terapia de substituição de gene se dá com a introdução de uma molécula, chamada de vetor, que carrega uma cópia normal de um gene. Os vetores geralmente são vírus, porque podem entrar em uma célula. No entanto, os vírus são projetados para não deixar as pessoas doentes. Alguns vetores comumente usados, chamados Vírus Adenoassociados, levam o gene normal para o núcleo da célula. Ao substituir o gene defeituoso (mutado) por uma nova cópia normal do gene, a célula agora produz uma proteína normal em vez da proteína anormal causadora de doenças.
Atualmente, a terapia de substituição de genes usando um vetor viral padrão não é viável para as doenças relacionadas ao RYR-1, porque o gene RYR1 é muito grande para ser empacotado nos vetores virais comumente usados. A terapia de substituição de genes poderá se tornar uma opção terapêutica no futuro, quando outros vetores paraentrega de genes poderão serem descobertos, ou quando técnicas totalmente novas poderão ser inventadas.
RYR-1 Foundation Clinical Care Guidelines
Lembre-se de que um gene é um segmento de DNA, um código de instruções sobre como produzir proteínas importantes. O DNA é uma molécula de fita dupla muito longa que tem uma forma helicoidal torcida, como uma escada sinuosa. Os blocos de construção do DNA são chamados de nucleotídeos. Existem quatro tipos de nucleotídeos: adenina (A), citosina (C), guanina (G) e timina (T). Uma importante coisa a lembrar é que a sequência de nucleotídeos (A,C,T e G) é a instrução que suas células usam para produzir proteínas. Na maioria dos casos, as doenças genéticas graves são causadas por um único erro ortográfico deste código, por exemplo, você tem um "A" onde deveria haver um "G". Esses erros no código de DNA podem impedir que suas células produzam uma proteína importante ou uma versão ruim disso. O objetivo da edição genética é corrigir permanentemente essas mudanças, alterando a sequência do seu próprio DNA. Pense em levar uma borracha e um lápis para corrigir um erro de ortografia em uma carta manuscrita. Em princípio, isso parece fácil, mas ainda é um grande desafio de fazê-lo de forma correta, eficiente e segura em todas as células.
A maioria das abordagens atuais de edição de genes envolve o uso de proteínas chamadas nucleases as quais cortam o DNA em locais específicos. Em geral, você precisaria de cortar o DNA apenas no local ou próximo ao nucleotídeo incorreto. Pense em colocar a borracha exatamente na palavra que você precisa reescrever. Você não gostaria de cortar DNA em outros lugares, porque isso poderia causar erros perigosos que mudariam o significado do código e fariam com que suas células funcionassem incorretamente.
A mais comumente nuclease usada para edição de genes é o sistema CRISPR/Cas9. Esta técnica é composta por duas partes: Cas9, que é uma proteína que foi descoberta em bactérias, e um RNA Guia. Cas9 e o RNA Guia se unem para formar a nuclease ativa, que você pode imaginar como borracha para a carta manuscrita. Essas tesouras se ligarão ao DNA com base na sequência do RNA Guia. Os cientistas podem alterar a sequência do RNA Guia conforme necessário para cortar quase qualquer sequência de DNA, com algumas limitações. Uma vez que o DNA é cortado, ele será reparado pela célula. Controlar o processo de reparo do DNA determina quais tipos de mudanças podemos fazer ao DNA. Ao contrário da edição precisa em uma carta manuscrita, ainda não temos controle sobre como a sequência será alterada. Alguns tipos de mudanças são fáceis de fazer, enquanto outros são mais difíceis e menos previsíveis.
Além de CRISPR/Cas9 e outras nucleases de edição de genes (por exemplo, dedos de zinco), também existem novas ferramentas promissoras em desenvolvimento. “Editores de base” são versões modificadas do sistema CRISPR/Cas9 que não cortam completamente a molécula de DNA de fita dupla. Em vez disso, eles cortam apenas uma fita, e mudam quimicamente um nucleotídeo para outro dentro de uma determinada janela de edição. Pense como mudar um “l” para um “t” adicionando um traço horizontal a um documento manuscrito. Nenhuma borracha foi necessária, já que esta versão do Cas9 não corta completamente o DNA. Portanto, o risco de falta de palavras é minimizado. A eficácia de editores de base está melhorando e eles têm vantagens significativas para corrigir alterações de nucleotídeo único. No entanto, mais trabalho é necessário para melhorar sua precisão.
A adição mais recente à caixa de ferramentas de edição de genes é o Prime Editing, ou Sistema de Edição Principal. A vantagem de “Prime Editors” é que eles nos deixariam corrigir praticamente qualquer tipo de pequeno erro no DNA, que atualmente não é possível com editores de base. Por exemplo, você pode modificar qualquer letra do alfabeto para qualquer outra, não apenas convertendo “l” para “t” (como no exemplo anterior). No entanto, muito mais trabalho é necessário para demonstrar que eles são seguros, eficazes e confiáveis.
Para resumir, a Edição de Genes é uma tecnologia muito empolgante que só recentemente se tornou possível, principalmente em um ambiente de laboratório de pesquisa. Em contraste com a terapia gênica tradicional, o objetivo é precisamente mudar o DNA do próprio paciente em cada célula do tecido afetado. Isso seria garantir que as mudanças forneçam um conjunto preciso e permanente de instruções de DNA que duraria por toda a vida. Embora esse campo seja uma grande promessa, as ferramentas atuais ainda têm grandes limitações, sendo que um dos maiores é entregar o mecanismo de edição do genoma a intencionada célula alvo do corpo do paciente. Outras considerações incluem o controle de qualquer resposta imune a impedir que o corpo de um paciente ataque as ferramentas de edição de genes. Além disso, maior precisão é necessário para controlar os tipos de alterações a serem feitas no local pretendido (no alvo edição), bem como evitar erros em outras partes do DNA de um paciente (edição fora do alvo). Apesar desses desafios, o futuro da edição de genes é muito brilhante e é algo para se esperar nos próximos anos.
O diagnóstico de uma doença rara impõe ao indivíduo afetado além das consequências físicas a ela inerentes, alterações na sua rotina de vida. Esse mesmo diagnóstico pode pôr fim a um período de vida marcado por incertezas e ansiedades, muitas vezes em razão de uma demorada e sofrida peregrinação por consultas e exames médicos. Entretanto, com o diagnóstico em mãos começa-se uma nova etapa, que é a busca por respostas acerca da doença em questão, e que muitas vezes o médico não conseguiu responder. E o maior desafio em meio a essas buscas se dá por um tratamento eficiente visando a cura, ou ao menos de um que seja pelo alívio do seu sofrimento físico. Portanto, para o paciente essa dura realidade do diagnóstico, é normalmente dividida em duas etapas, uma que acontece no consultório médico, e a outra etapa, que se vive dentro das "quatro paredes da sua intimidade". O processo de assimilação do diagnóstico pode ser assustador e cheio de interrogações, e o indivíduo afetado tende buscar por si só as respostas que mais anseiam, tais como sobre prognóstico, cura, e tratamento. Essas buscas e pesquisas são normalmente feitas na internet, contudo, muitas vezes ao contrário de esclarecer a situação, pode torná-la mais confusa.
Entendo perfeitamente a reação desses indivíduos afetados, assim como de seus familiares, uma vez que o desconhecimento por muitos médicos sobre essas doenças ainda é bem limitado. Enfim, na prática, uma vez diante do diagnóstico que você é portador de uma doença que nunca havia ouvido falar a respeito anteriormente, e que ouve do médico, pessoa esta que se imaginava ser a quem poderia lhe ajudar, lhe dizer que a tal doença é rara, que se tem muito pouca informação sobre ela, e que para a mesma não tem cura e nem mesmo tratamento eficaz, e que seu prognóstico é de progressividade, …daí você como parte interessada, o que lhe resta ? … além das buscas na internet por informações, o maior desejo é encontrar outras pessoas que estejam vivendo com o mesmo diagnóstico. O SORRYR-1 tem se prestado a este propósito, tanto que tenho sido procurado por indivíduos afetados e/ou por seus familiares questionando sobre o que tenho feito para lidar com a doença nestes meus 60 anos de vida. Nas primeiras vezes em que fui questionado, confesso que tive dificuldade em responder, primeiro porque entendo que cada pessoa é única, e o que funciona para uma pessoa pode não funcionar para outra, mas com o tempo pude entender que o meu posicionamento pessoal poderia ser algo complementar ao que me propus com o SORRYR-1. Assim sendo, em atendimento a algumas sugestões, decidi escrever de maneira prática sobre o que entendo ser o mais importante para conviver com as doenças relacionadas ao RYR1, e especificamente no meu caso a Miopatia Congênita Centronuclear.
1 - Inicialmente falo sobre minha convicção pessoal dizendo que creio viver esta situação segundo um propósito, me baseando na palavra do apóstolo Paulo em Romanos 12:2 que diz, “.... transformem-se pela renovação da sua mente, para que sejam capazes de experimentar e comprovar a vontade de Deus para sua vida.” , entendendo que apesar das imposições a mim colocada pela doença, busco reinventar e viver um estilo de vida segundo minhas possibilidades, com espírito de resiliência e superação, e busco servir ao próximo como sendo este o propósito de Deus para minha vida.
2 - Atitudes positivas devem ser a palavra de ordem na sua maneira de viver. Assim, entender que o bem-estar emocional é fundamental para uma vida saudável e feliz, faz com que não deixe que sua condição física interfira no seu estado emocional. É importante também lembrar que todos enfrentamos desafios e dificuldades em diferentes aspectos de nossas vidas, e a deficiência física é apenas uma parte de quem somos. Portanto diante do espírito de resiliência e auto-estima positiva, busque se superar nas suas possibilidades e habilidades, não se limitando nas suas limitações, ao contrário, as potencializando.
3 - A atividade física é primordial, é nosso único tratamento disponível, seja fisioterapia ou ginástica, e essa prática deve ser parte de nossa rotina diária. Vale lembrar que nossas células musculares, pela mutação que temos no RYR1 não funciona da maneira correta (contração e relaxamento), interferindo assim em questões relacionadas ao nosso fortalecimento e movimentação, além de poder causar contraturas, rigidez, dores, fadiga, dentre outras. A atividade física, mesmo que passiva, pode nos ajudar com a qualidade de vida e até impedir a progressão da doença. Lançando mão da frase dita por meu neurologista, Dr Acary, que diz, “o RYR1 é movimento”, portanto, movimente, mexa-se, nunca pare de mexer, mexa do músculo do dedo do pé até os da face, assim, mexa-se sempre. Contudo, vale ressaltar que devemos estar atentos ao nosso limite, evitando o excesso de esforço físico e cansaço, pois isso pode agravar os sintomas da miopatia;
4 - O condicionamento respiratório também deve ser um grande ponto de atenção diante das dificuldades musculares generalizadas que enfrentamos. O enfraquecimento da musculatura do tórax, traqueia e diafragma pode interferir em questões sistêmicas. Assim, destaco como exemplo a dificuldade na expectoração de eventuais secreções, deglutição, e adequada troca de gases feita pelos pulmões. Leia mais sobre este tema na postagem “Abordagem Respiratória na Miopatia Centronuclear”;
5 - Outro ponto que deve ser observado é com nosso ganho de peso, uma vez que o quê se ganha nesa situação é a gordura, e como nosso tecido muscular que já não é lá essas coisas, ele é substituído com facilidade pelo tecido gorduroso. O aumento de peso dificulta ainda mais nossa capacidade em fazer atividade física, que por sua vez impede a queima de calorias, favorecendo assim ainda mais o acúmulo de gordura em nosso corpo. Esse acúmulo de gordura em nosso corpo, além de afetar nossa qualidade de vida, pode ainda nos trazer consequências patológicas graves, tais como cardiopatias, diabetes, hipertensão arterial, doenças no fígado, alguns tipos de câncer, problemas renais, dentre outros.
6 - Considerando nossa reduzida capacidade em fazer atividade física, temos uma propensão à fragilidade óssea, portanto, temos que evitar as quedas pelo alto risco de fraturas. Na prática, é que para uma fratura óssea, o tratamento deverá ser desde a imobilização do membro afetado, até a intervenção cirúrgica (observar riscos cirúrgicos), e em ambas situações ficaremos impedidos de movimentação física, o que causará ainda mais perda muscular.
7 - Evite contrair qualquer doença, seja um simples resfriado, ou qualquer outra doença. Por exemplo, nosso corpo diante de uma enfermidade com causa viral ou bacteriana, reage com uma resposta imunológica, liberando uma série de proteínas, as citocinas, as quais produzem uma reação inflamatória não apenas no local da infecção, mas também em outros órgãos, incluindo músculos e articulações, que são nosso maior ponto de atenção e fragilidade. Assim, com esse exemplo procurei mostrar porque uma simples gripe pode nos causar ainda mais fraqueza.
8 - Mantenha um estilo de vida praticando hábitos saudáveis, tais como, seguir uma dieta mais nutritiva, equilibrada, e saudável, ingerindo bastante água (35 ml/kg de peso corporal por dia), procure dormir o suficiente e com qualidade, se exponha periodicamente ao sol (8 às 11 horas), enfim, evite o estresse e preocupação excessiva, assim como hábitos nocivos a saúde física (cigarro, droga, e álcool).
9 - Fique atento e evite tratamentos experimentais, e medicamentos que não os indicados por seu médico especialista. Por exemplo, alguns medicamentos, como estatinas, anti-inflamatórios não esteroides (AINEs), e outros mais, podem piorar a miopatia.
10 - Faça regularmente um acompanhamento médico com especialista visando monitorar não somente sua miopatia, mas também sua condição física geral. É muito importante que os portadores de miopatia conversem com seu médico para discutir os fatores que podem piorar sua condição, e que possam aprender a gerenciá-los.
As Doenças Relacionadas ao RYR1 são consideradas como “doenças raras” que afetam os músculos esqueléticos. A mutação no gene RYR-1, responsável pelo desenvolvimento da patologia, tem um impacto significativo na vida das pessoas afetadas e de seus familiares. Além dos sintomas e limitações físicos, como fraqueza muscular, rigidez, dificuldades respiratórias, atrofia muscular, entre outros, a doença tende a afetar seriamente os aspectos psicológicos e emocionais.
Para indivíduos nessa condição, o medo pode se tornar paralisante e afetar a qualidade de vida de diversas maneiras. O medo da morte pode ser comum, visto que a doença não tem cura, e a incerteza sobre a evolução da patologia pode gerar insegurança e ansiedade. Além disso, o medo de como será visto, julgado e excluído pode surgir como consequência da falta de conhecimento e sensibilidade da sociedade em relação às doenças raras.
O medo de atrapalhar as pessoas em função da limitação física ou dependência emocional também pode afetar a qualidade de vida das pessoas portadoras das doenças relacionadas ao RYR1 e seus familiares. Esse medo pode levar ao isolamento social e à falta de participação em atividades cotidianas, o que pode agravar ainda mais os sintomas físicos e emocionais da doença.
Contudo, um dos fatores que merece alerta em relação ao medo é que ele pode afetar negativamente a busca por tratamento, já que a falta de informação e resultados eficazes tende a causar ansiedade e frustração diante de promessas de tratamentos sem resultados efetivos e diretos na causa da doença. O medo pode gerar limitações (barreiras) na adesão ao tratamento, uma vez que a falta de confiança pode levar à desistência ou ao abandono das práticas recomendadas.
É comum também que a pessoa, frente a esse cenário, tenda a desenvolver um mecanismo inconsciente de defesa e comece a se convencer de que não vale a pena aderir ao tratamento, esse é um processo inconsciente para diminuir a dor das constantes frustrações. Então, para não se frustrar ainda mais e evitar decepcionar os familiares, ela sabota o tratamento, no fundo ela teme não conseguir. A falta de conhecimento também pode gerar preconceitos e discriminação por parte da sociedade, o que pode dificultar ainda mais o enfrentamento da doença.
Portanto, é importante trabalhar na conscientização e na superação desses medos, para que o tratamento, mesmo que este tratamento não seja curativo, mas de manutenção, mas seja assim, eficaz e a qualidade de vida seja conquistada. A conversa clara e aberta, como estamos tendo aqui é fundamental para o enfrentamento da realidade. A colaboração de profissionais da psicologia e familiares é fundamental para apoiar a pessoa a lidar com esses medos e com a ansiedade e assim encontrar as melhores soluções para superá-los. Faz sentido para você? E no próximo texto vamos falar sobre a melhor forma de conseguir os resultados psicológicos necessários aqui citados.
O texto acima foi escrito e gentilmente cedido ao SORRYR-1 como colaboração pela Psicóloga Mirella Nery (CRP 09/002785) - @mirellanerypsi
Dado à seriedade deste assunto, devo iniciar o texto com as palavras conclusivas sobre o tema… a HIPERTERMIA MALIGNA é uma doença grave, com risco de morte, que se apresenta em forma de crise, acionada por um gatilho, que é normalmente uma droga anestésica, sendo os portadores de mutação no gene RYR1, os indivíduos com maior risco de ser afetado, daí a afirmação, “HIPERTERMIA MALIGNA, PONTO DE ATENÇÃO PARA OS MÉDICOS ANESTESISTAS, PREOCUPAÇÃO PARA OS PORTADORES DE MUTAÇÃO DO GENE RYR1”. Os anestesistas devem ter o conhecimento sobre a doença e assumir que todas as pessoas com mutação no gene RYR1 correm risco de Hipertermia Maligna. No caso de qualquer procedimento médico que seja necessário anestesia, o portador de mutação de no gene RYR1 deve fazer com que o cirurgião e anestesista saiba sobre sua mutação no gene RYR1, portanto com Suscetibilidade a Hipertermia Maligna (MHS), assumindo assim os eventuais riscos, para tomar precauções e administrar um tipo seguro de anestésico. É recomendado também aos portadores da mutação do RYR1 que portem uma identificação de advertência médica indicando seu risco de HM, em caso de emergência, neste caso, um adesivo em todos os documentos pessoais (Ex.: Identidade, CNH, Passaporte, Carteira do Plano de Saúde, e etc).
CONCEITO
A Hipertermia Maligna (HM) é uma patologia de relação farmacogenética. E isso significa que a doença se manifesta através de um episódio/crise em indivíduos que tenham uma suscetibilidade genética, devido a uma mutação em um determinado gene, acontecendo caso sejam expostos a gatilhos anestésicos (fármacos = medicamentos). Essa predisposição é chamada de "Suscetibilidade à Hipertermia Maligna” (MHS). Genericamente, explica-se que a Hipertermia Maligna (HM) é uma reação biológica em que o corpo humano superaquece a ponto de um colapso muscular, e é considerada uma emergência médica. Se alguém com Hipertermia Maligna não for tratado a tempo, como consequência pode resultar em insuficiência renal, dano cerebral, parada cardíaca, falência de órgãos adicionais, e até morte.
Os indivíduos com Suscetibilidade à Hipertermia Maligna (MHS) podem também apresentar uma crise em resposta a outros gatilhos externos, como por exemplo, ao esforço físico, que poderá causar a Rabdomiólise (quebra muscular), e neste caso apresentando outros sintomas, tais como, cãibras severas, rigidez muscular, e intolerância ao calor.
A genética à Suscetibilidade à Hipertermia Maligna (MHS) é complexa, e vários genes têm sido identificados desempenhando um papel patogênico na Hipertermia Maligna, sendo o RYR1 o mais estudado. Este gene codifica o receptor 1 de Ryanodina (RyR-1), uma proteína do canal de cálcio do retículo sarcoplasmático, expressa predominante na célula muscular. Sabe-se que na maioria dos casos da Suscetibilidade à Hipertermia Maligna (MSH), ela apresenta um traço autossômico dominante, e isso significa que se você tem MHS, um de seus pais provavelmente também tem a MHS. Também significa que cada um de seus filhos têm 50% de chance de herdar a MHS. No entanto, a dita complexidade se prova quando os médicos também observaram a Hipertermia Maligna em pessoas com mutações RYR1 autossômicas recessivas.
Em alguns casos, ao contrário do que se pensa, a Suscetibilidade à Hipertermia Maligna (MHS) pode ocorrer na ausência de fraqueza muscular, em outras palavras, os indivíduos com MHS têm força normal ou até mesmo aumentada, e seu único “sintoma” é a suscetibilidade a reações de Hipertermia Maligna (HM). Por outro lado, a MHS também pode ocorrer em pacientes com Doenças Relacionadas ao RYR1 (RYR-1-RD) com sinais e sintomas típicos de miopatia (fraqueza muscular). Os gatilhos para Hipertermia Maligna incluem certos medicamentos usados para anestesia geral, ou seja, quando alguém é “colocado para dormir”, geralmente antes de uma cirurgia. A anestesia geral é usada em uma ampla variedade de ambientes, incluindo salas de cirurgia, salas de emergência e unidades de terapia intensiva (UTI). Medicamentos específicos conhecidos por desencadear a Hipertermia Maligna incluem anestésicos administrados por via intravenosa, e inalatória via tubo respiratório, como segue:
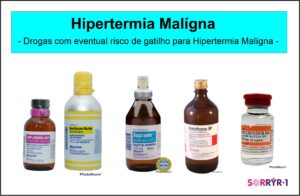
Mutações no RYR1 associadas a Hipertermia Maligna (HM) mostram uma penetrância variável, e isso significa que uma pessoa pode passar por várias exposições a gatilhos sem problemas antes que uma reação de HM ocorra pela primeira vez. Para tornar as coisas ainda mais confusas, as pessoas com a mesma mutação (incluindo membros da mesma família) podem ter reação clínica diferente, o que significa que algumas podem ser sensíveis ao calor, algumas podem ter reações de HM à anestesia, algumas podem ter rabdomiólise com exercícios físicos, e algumas podem não ter problemas com nenhuma dessas condições.
QUADRO CLÍNICO
O quadro clínico de um episódio/crise de Hipertermia Maligna é variável, e compreende manifestações de alterações metabólicas, de lesão muscular, e das complicações secundárias. Esta condição é expressa por rigidez muscular, aumento do consumo de oxigênio e produção de gás carbônico, acidemia (respiratória e metabólica), taquicardia, taquipnéia, hiperpotassemia, rabdomiólise e mioglobinúria. A dessaturação da hemoglobina no sangue arterial pode ser identificada por oximetria de pulso. Entre os diversos fatores que potencialmente contribuem para a dessaturação persistente, encontram-se acidemia, hipercarbia e hipertermia, capazes de deslocar a curva de saturação da hemoglobina para a direita. A hipercarbia, já detectada na cartografia, parece preceder as demais manifestações. A forma fulminante da Hipertermia Maligna é caracterizada por hipercapnia, rigidez muscular, hipertermias graves, e rabdomiólise, mas situações como cirurgias cardíacas sob circulação extracorpórea (CEC) com hipotermia podem atenuar a expressão clínica da Hipertermia Maligna (HM). A hiperventilação pode mascarar o diagnóstico de HM. Bloqueadores neuromusculares podem retardar o início das manifestações da crise de HM. Convém destacar que nem sempre hipertermia é manifestação inicial ou proeminente da HM. A rigidez muscular pode inexistir em 25 % dos casos, e a Hipertermia ser registrada em apenas um terço deles. A HM surge a qualquer momento durante a anestesia, tendo sido descrita sua ocorrência até 3 horas após a interrupção da exposição ao agente desencadeante (gatilho). A crise de Hipertermia Maligna (HM) pode manifestar-se tardiamente, mesmo após a interrupção da administração do agente desencadeante (gatilho), talvez a imobilidade determinada pela própria anestesia limite a liberação de cálcio a partir do retículo sarcoplasmático. Ao acordar, aumenta a atividade muscular e, na presença de resíduos anestésicos, vêm-se potencializadas à liberação intracelular de cálcio e seus efeitos metabólicos. Tem-se a impressão de haver diferenças entre os halogenados com relação ao seu potencial para desencadear crises de Hipertermia Maligna (HM). O halotano parece ser o de maior risco. A exposição ao isoflurano pode associar-se à crise de HM de início tardio. Parece que a indução da liberação de cálcio do retículo sarcoplasmático pelo sevoflurano é menos intensa em comparação aos demais agentes.
Observação de Manifestações Clínicas Iniciais: Taquicardia - 96,0%, Rigidez muscular - 83,6%, Instabilidade hemodinâmica - 85,5%, Taquipnéia - 85,0%, Cianose - 71,1%, Hipertermia - 30,0%
TERAPIA
Dada a gravidade da doença, o tratamento da Hipertermia Maligna no regime operatório deve ser iniciado em caráter de emergência. A administração do anestésico deve ser interrompida imediatamente, e o paciente deve então receber infusão intravenosa de dantroleno sódico, um relaxante muscular que restaura os níveis fisiológicos de cálcio nos músculos.
Posteriormente, o paciente é induzido a baixar a temperatura corporal por meio de fluidos frios e bolsas de gelo para evitar consequências no cérebro, e é administrado oxigênio para satisfazer o aumento da demanda do organismo. Além disso, a acidose metabólica induzida por lactato é prontamente tratada e o desequilíbrio eletrolítico corrigido. O sucesso da intervenção depende em grande parte da rapidez no reconhecimento dos sintomas e da resposta individual do paciente à terapia.
Nos últimos trinta anos, graças a novos estudos e descobertas no campo farmacológico, a taxa de mortalidade da hipertermia maligna caiu drasticamente, de 70-80 % para 5%, tornando-se uma doença relativamente manejável e tratável.
ESPERANÇA AOS PORTADORES DE DOENÇAS RELACIONADAS AO RYR1 VISTA POR UM OUTRO LADO DO PRISMA
Na postagem anterior tratei sobre as boas notícias que tive durante o workshop científico sobre as doenças relacionadas ao RYR1 em Pittsburgh (EUA), em julho de 2022. Me referi às pesquisas sobre uma droga capaz de aliviar, tratar, e até curar as doenças relacionadas ao RYR1, mencionei inclusive sobre valores de investimentos em pesquisas e tendências de mercado para os próximos anos. Contudo pode ter ficado uma pergunta no ar, sobre o porquê do grande interesse em pesquisas tão dispendiosas sobre uma doença que atinge um pequeno número de pessoas. …então vamos buscar o raciocínio lógico…
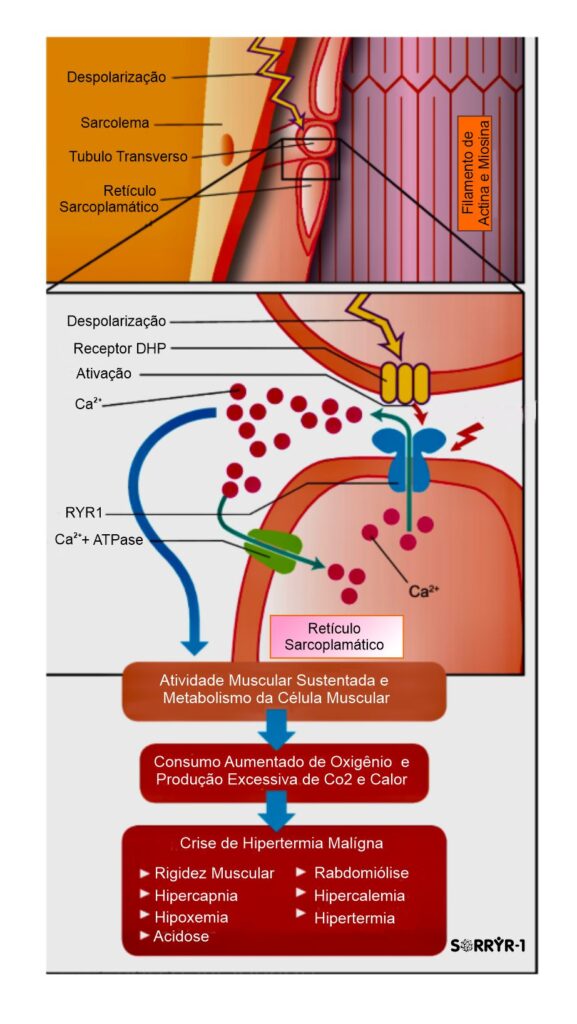
Músculo é o tecido responsável pelo movimento do corpo humano. O mecanismo de funcionamento do músculo, ou seja, como ele contrai e relaxa, é chamado de acoplamento excitação-contração (EC), e tudo acontece na célula da fibra muscular, onde o citoplasma está localizado. Lá dentro, o Retículo Sarcoplasmático (RS) atua como reservatório para íons de cálcio, essenciais para desenvolver a força muscular. O receptor de Ryanodina Tipo 1 (RYR1) funciona como canal liberador dos íons de cálcio, o qual quando liberado no citoplasma, faz com que o músculo se contraia. Na prática o mecanismo de funcionamento é simples, quanto mais íons de cálcio liberado no citoplasma, mais força muscular o indivíduo desenvolve.
O cientista Andrew R. Marks, MD, professor de fisiologia e biofísica celular na Universidade de Columbia (EUA), explica em seus estudos que a perda da função muscular é associada à disfunção dos canais de liberação do receptor de rianodina (RYR1) do músculo, e que pode ser causado por uma falha decorrente a uma mutação genética, ou devido à sobrecarga oxidativa celular relacionada à idade. Essa falha desestabiliza o estado fechado do canal, resultando em um defeito pelo vazamento de cálcio intracelular, acarretando em função muscular reduzida, o que explica não somente as doenças relacionadas ao RYR1, mas também explica no caso da oxidação celular, a razão pela qual os exercícios físicos se tornam mais difíceis com a idade.
A boa notícia é que existe uma droga em fase de testes que pode barrar esse vazamento. Na pesquisa foram estudadas as células musculares de ratos novos e velhos. Um rato de seis meses cujos receptores de rianodina vazavam cálcio mostraram os mesmos problemas de fraqueza muscular do que um rato mais velho. Este, por sua vez, apresentou melhoras depois de ser tratado com a droga em teste. O estudo sugere aos cientistas procurarem por novos caminhos no tratamento do envelhecimento. “As pesquisas que temos visto se focam em produzir mais músculos”, diz Andrew Marks. “E a diferença é que nós focamos não no músculo, mas no seu mecanismo de funcionamento, pois o aumento de músculos não ajuda se eles não funcionarem.”
Portanto, entendo que quando a comunidade científica desenvolve pesquisas em busca de uma droga capaz de tratar e até curar as doenças musculares relacionadas ao RYR1, se busca também beneficiar todos os indivíduos, porque a fraqueza muscular decorrente do envelhecimento é comum a todos. Assim, podemos pensar que o interesse mercadológico para a descoberta de uma droga capaz de atuar no defeito ou mal funcionamento da célula muscular, pode ser maior por ir de encontro com a necessidade de todos os indivíduos, e não somente aos portadores de doenças relacionadas ao RYR1. Por fim eu faço a seguinte analogia, nós portadores de uma doença relacionada ao RYR1 temos um defeito de fábrica, que é a mutação genética, e os indivíduos saudáveis terão um defeito por conta do tempo ou idade, que é a oxidação celular.

