O gene RYR1, descrito em 1989 por MacLennan é responsável por dar as instruções para a proteína do receptor de rianodina 1 (RYR1), que é um canal de cálcio crucial para a contração e relaxamento dos músculos esqueléticos.1 A função principal do gene é codificar o canal que libera íons de cálcio do retículo sarcoplasmático (um reservatório dentro da célula muscular) quando o músculo é ativado por um sinal elétrico. Quando o canal RYR1 se abre, o cálcio entra no citoplasma, permitindo que os músculos se contraiam para permitir o movimento. Variantes (mutações) neste gene podem levar a doenças musculares como miopatias congênitas: Doença do Núcleo Central (Central Core), Doença Multi-Minicore, Miopatia Centronuclear e Desproporção Congênita do Tipo de Fibra. O estudo destas doenças permitiu o sequenciamento do gene RYR1 e a descoberta da relação de variantes neste gene e manifestações clínicas como Hipertermia Maligna (HM), Rabdomiólise e Rabdomiólise por Esforço (RE).
RYR1 está localizado no cromossomo 19q13.1 e codifica o Receptor de Rianodina Tipo 1 (RYR1). O gene RYR1 compreende 106 exons e codifica uma grande proteína com 5038 aminoácidos. Devido ao seu tamanho e complexidade consideráveis, o RYR1 tem sido historicamente um gene desafiador para estudo. Durante muitos anos, os esforços de pesquisa concentraram-se principalmente em três domínios (N-terminal, central e C-terminal) considerados pontos críticos de mutação. No entanto, com o advento das tecnologias de Sequenciamento de Nova Geração (NGS), tornou-se possível analisar o gene inteiro. Como resultado, inúmeras novas variantes foram identificadas fora dos pontos críticos previamente conhecidos. Até o momento, mais de 1000 variantes distintas do gene RYR1 foram relatadas (HGMD; https://www.hgmd.cf.ac.uk/ (acessado em 04 de dezembro de 2025) e LOVD; https://databases.lovd.nl/shared/genes/RYR1 (acessado em 04 de dezembro de 2025), porém apenas 72 foram classificadas como variantes diagnósticas pelo Grupo Europeu de Hipertermia Maligna (EMHG; https://www.emhg.org/diagnostic-mutations (acessado em 04 de dezembro de 2025)). A grande maioria dessas variantes são de sentido trocado, enquanto inserções, deleções e duplicações são relativamente incomuns, representando menos de 10% de todas as variantes patogênicas conhecidas no gene RYR1.
A Hipertermia Maligna (HM) é uma síndrome hipermetabólica farmacogenética que se manifesta como uma crise aguda após a exposição de indivíduos suscetíveis a agentes halogenados e/ou succinilcolina. A crise aguda pode se manifestar clinicamente com hipertermia, hipercapnia, taquipneia, taquicardia, acidose metabólica, hipercalemia, rigidez muscular e rabdomiólise, sendo uma condição extremamente grave. Ocorre em pacientes de todas as etnias e distribuições geográficas, sendo duas vezes mais comum nos homens e menores de 19 anos de idade (50% dos casos). Se não tratada adequadamente, óbito ocorre em 80%–90% dos pacientes. O tratamento consiste em evitar as crises e abordagem seguindo protocolos rígidos nas crises agudas, incluindo-se uso de dantrolene de sódio (reduz a liberação de cálcio do retículo sarcoplasmático do músculo estriado esquelético por limitar a ativação do receptor de rianodina RYR1).2,3
A prevalência da crise de HM é variável, de 1:10.000 em crianças a 1:50.000 em adultos. A frequência relacionada a procedimentos anestésicos varia de 1 em 10.000 a 1 em 250.000, dependendo da população e dos critérios diagnósticos. A suscetibilidade à HM tem sido associada a genes ligados ao metabolismo do cálcio e que codificam proteínas do complexo de acoplamento excitação-contração do músculo esquelético, com uma frequência de 1:217–1:2750 na população geral. O principal gene implicado na suscetibilidade à HM é o RYR1, responsável por aproximadamente 75% dos casos geneticamente confirmados. Famílias raras apresentam variantes nos genes CACNA1S (1%), STAC3 (<1%) ou ASPH.
A apresentação clínica entre portadores de variantes do RYR1 é altamente variável, e o panorama genético é marcado por alta heterogeneidade alélica, penetrância incompleta e expressividade variável. Variantes patogênicas nesse gene resultam em liberação desregulada de cálcio do retículo sarcoplasmático, levando à contração muscular sustentada e crise metabólica.
Devido à heterogeneidade genética e à possibilidade de herança poligênica, o padrão ouro no diagnóstico de suscetibilidade à HM é o teste de contratilidade in vitro fenotípica (IVCT) para o grupo europeu ou o teste de contratilidade com cafeína-halotano (CHCT) para o grupo norte-americano. Atualmente, apenas um teste de contratilidade negativo pode excluir a suscetibilidade à HM, enquanto um teste genético negativo não o faz. No entanto, esforços têm sido feitos para melhorar a capacidade de detecção do teste molecular, que poderia ser uma ferramenta diagnóstica menos invasiva. Por outro lado, variantes no gene RYR1 são muito frequentes na população, e a identificação e classificação dessas variantes como patogênicas requerem testes adicionais e curadoria. Apenas 72 variantes patogênicas ou provavelmente patogênicas no RYR1 são reconhecidas de acordo com a lista do Grupo Europeu de Hipertermia Maligna (EMHG) ( https://www.emhg.org/genetic-scoring-matrix ; acessado em 18 de agosto de 2025), e, além dos critérios do EMHG, existem os critérios do painel de especialistas em curadoria de variantes do ClinGen MHS (VCEP). Variantes missense são as mais comuns, enquanto inserções e duplicações representam menos de 10%. Os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína.
Cada país ou região apresenta diferenças no tipo e na frequência de variantes associadas à HM. A frequência de variantes relatadas em pacientes com HM varia de 37% a 87,5%, sendo as variantes RYR1 p.R614 e p.G2434R as mais frequentes. No entanto, não existem dados genéticos populacionais da América do Sul, exceto pela descrição de casos isolados e famílias. Em estudo recente, desenvolvido na Unifesp – Escola Paulista de Medicina, foram revisados os dados clínicos e laboratoriais de todas as famílias encaminhadas para avaliação na Unidade Brasileira de HM devido a histórico pessoal ou familiar de HM durante anestesia. Foram coletados dados demográficos e clínicos, bem como níveis séricos de creatina quinase (CK), resultados do teste de contratilidade in vitro (TCIV) e resultados de estudos anatomopatológicos do músculo esquelético. A análise molecular foi realizada por meio de sequenciamento de exoma completo (NGS). Pacientes com e sem variantes foram comparados. Variantes no gene RYR1 foram encontradas em 38 pacientes (62,2%), e nenhuma variante foi identificada em 20 pacientes (32,7%). Mais de uma variante no RYR1 foi encontrada em seis indivíduos. Variantes no gene CACNA1S foram encontradas em três pacientes (4,9%), todos com variantes concomitantes no RYR1. Três pacientes apresentaram variantes no gene STAC3 (4,9%). Comparando os grupos de pacientes com variantes no RYR1 com o grupo sem variantes nesse gene, observou-se que o primeiro grupo apresentou valores séricos mais elevados de CK, maior frequência de ptose, estrabismo, e maior amplitude de contratura no TCIV após a administração de cafeína ou halotano. Nesta avaliação preliminar de indivíduos brasileiros com histórico de hipertermia maligna, a frequência de variantes no RYR1 foi semelhante à de relatos anteriores em outros países, porém houve maior frequência de variantes nos genes STAC3 e CACNA1S.4
Também em estudo da Unifesp, foi identificada uma variante rara: duplicação no gene RYR1 na variabilidade do fenótipo de susceptibilidade à Hipertermia Maligna, em uma família com dois irmãos afetados portadores de uma inserção de 18 pares de bases no éxon 91 do gene RYR1, resultando em uma duplicação em fase de 6 aminoácidos (c.12835_12852 dupGAGGGCGCGGCGGGGCTC: 162 p.G4279_T4284insAAGLEG). A expressão relativa do mRNA do gene RYR1 no músculo dos dois pacientes identificou uma redução de aproximadamente 50%, sugerindo um possível alelo hipomórfico. Este achado levanta a questão que os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína. Este estudo reforça a possibilidade de que a duplicação nessa região possa causar defeitos estruturais e um fenótipo mais grave nos pacientes.5
A Rabdomiólise é uma condição potencialmente fatal que envolve a rápida dissolução do músculo esquelético em resposta a uma variedade de fatores desencadeantes, clinicamente caracterizada por um aumento súbito e acentuado, seguido de uma queda nos valores séricos de creatina quinase (CK) As causas mais comuns de rabdomiólise são lesões por esmagamento secundárias a traumas, esforço físico extremo e miopatias metabólicas. As principais características incluem dor muscular e uma elevação súbita e transitória dos valores séricos de CK. A rabdomiólise grave é frequentemente acompanhada por aumento da excreção urinária de mioglobina (mioglobinúria), o que pode levar à insuficiência renal aguda (IRA) e a uma crise metabólica potencialmente fatal. A ampla gama de complicações (por exemplo, insuficiência renal aguda, arritmias cardíacas, síndrome compartimental, coagulação intravascular disseminada) enfatiza a relevância clínica da rabdomiólise em diversas especialidades médicas.
Estudos de coorte retrospectivos focados em rabdomiólise em pacientes hospitalizados na era pré-sequenciamento de nova geração (NGS) concentraram-se particularmente em fatores desencadeantes externos como a principal causa de um evento de rabdomiólise. Esses estudos identificaram toxinas exógenas (drogas ilícitas, álcool), trauma muscular direto, infecções e exercícios extenuantes (Rabdomiólise por Esforço) como alguns dos fatores desencadeantes mais comuns. Exames genéticos de última geração têm associado mais de 30 genes a uma maior suscetibilidade à rabdomiólise. Contudo, um desafio fundamental na abordagem diagnóstica reside na consideração de quais pacientes necessitam de triagem genética diagnóstica após um episódio de rabdomiólise para identificar uma doença neuromuscular ou metabólica pauci-sintomática ou assintomática. Dentre os genes relacionados, destaca-se o RYR1 e variantes nele podem ser responsáveis por uma proporção substancial de pacientes que apresentam sintomas inexplicáveis de rabdomiólise e/ou mialgia por esforço.6
Com o intuito de revisar a abordagem diagnóstica genética da rabdomiólise, uma pesquisa na Holanda foi realizada, tendo como palavra chave o acrônimo 'RHABDO': Recurrent episodes (episódios recorrentes); HyperCKaemia persisting 8 weeks after the event (aumento de CK persistente após 8 semanas do evento); Accustomed exercise—the intensity of the exercise cannot sufficiently explain the rhabdomyolysis event (exercício de costume – a intensidade do exercício não explica por si o evento de rabdomiólise); Blood CK > 50× the upper limit of normal (ULN) or >10,000 IU/L in female Caucasian patients (aumento de CK > 50x ou > 10 000 UI/L em mulheres); Drugs/medication and other exogenous triggers are insufficient to explain the event (drogas/medicações e outros agentes exógenos são insuficientes para explicarem o evento); and Other affected family members or other exertional symptoms (e.g., severe muscle cramps or swelling) (outros familiares afetados ou com sintomas relacionados ao exercício físico). O acrônimo foi baseado em uma revisão da literatura e em nossa experiência clínica e, portanto, atingiu o nível de evidência de opinião de especialistas. A relevância do RHABDO é ainda mais reforçada por um recente workshop do Centro Neuromuscular Europeu (ENMC) envolvendo 21 médicos e pesquisadores de 12 países diferentes, que enfatizou a necessidade de pesquisa coordenada nesta área. Neste estudo retrospectivo bicêntrico, 122 pacientes foram incluídos. Os fatores desencadeantes mais frequentemente relatados que contribuíram para eventos de rabdomiólise foram exercício (72%), febre/infecção (22%) e/ou medicação (18%). Eles foram submetidos a avaliação genética através de painéis genéticos relacionados à miopatia metabólica (82%), sequenciamento de Sanger (49%) e sequenciamento de exoma completo (NGS) (24%), dos quais 52 pacientes (43%) foram submetidos a múltiplos métodos. Uma variante (provavelmente) patogênica foi identificada em 13 pacientes (11%), todos com ≥2 características de RHABDO presentes. O valor preditivo positivo para ≥2 características foi de 14%, enquanto o valor preditivo negativo foi de 100%. Variantes no gene RYR1 foram descritas em quatro pacientes, um relacionado com esforço (RE).7
A Rabdomiólise por Esforço (RE) é uma degradação muscular patológica associada à atividade física extenuante e agravada por múltiplos fatores de risco. Estes incluem baixo nível de condicionamento físico, alto índice de massa corporal, infecção viral em curso e altitude e temperatura elevadas. A sua incidência é de aproximadamente 36,5 por 100.000 pacientes-ano em atletas ou uma taxa semelhante em militares. De modo geral, as diferenças fundamentais entre a RE e outras formas de rabdomiólise residem em suas causas, fatores desencadeantes e populações afetadas. A patologia celular da lesão por esforço repetitivo (LER) centra-se na ruptura da integridade das células musculares, particularmente no que diz respeito à homeostase iônica e à produção de energia. Exercícios extenuantes ou incomuns podem causar lesão direta ao sarcolema e/ou levar à falha na produção de energia, comprometendo a função de bombas iônicas essenciais, como a Na + /K + -ATPase e a Ca2 + -ATPase. Esse comprometimento aumenta a permeabilidade celular aos íons sódio, resultando em um influxo significativo de cálcio (Ca2 +) para as fibras musculares. O aumento da concentração intracelular de cálcio ativa enzimas dependentes de cálcio, incluindo proteases e fosfolipases, que iniciam a destruição de proteínas miofibrilares, citoesqueléticas e de membrana. Esse processo leva à necrose das fibras musculares, liberando conteúdos intracelulares como CK, mioglobina e eletrólitos no fluido extracelular e na circulação sanguínea. O ciclo vicioso resultante envolve contração muscular sustentada devido ao aumento do cálcio, o que esgota ainda mais as reservas de energia e exacerba o dano muscular.
Embora a via celular geral envolvendo sobrecarga de cálcio e depleção de energia, seja compreendida como o mecanismo de dano às células musculares, os mecanismos subjacentes específicos da rabdomiólise (RB) não são universalmente compreendidos. Estudos em modelos animais, como cavalos suscetíveis à RB recorrente, utilizaram com sucesso a análise do transcriptoma (RNA-seq ou análise de microarray) para revelar alterações na expressão gênica em vias relacionadas à regulação do cálcio, estresse oxidativo e função mitocondrial, demonstrando que essas alterações moleculares podem persistir mesmo entre os episódios. No entanto, as fontes fornecidas não oferecem uma visão abrangente de pesquisas semelhantes em larga escala sobre o transcriptoma, conduzidas especificamente em populações humanas com RB. Um estudo elegante com sequenciamento de RNA em amostras de músculo esquelético de 19 pacientes humanos com histórico de RE, coletadas no mínimo seis meses após o evento de RE mais recente, e oito controles saudáveis para investigar o perfil transcriptômico da RE revelou uma forte supressão da função mitocondrial. Essa supressão incluiu as vias da “cadeia de transporte aeróbico de elétrons” e da “fosforilação oxidativa”, indicando comprometimento da produção de energia. Por outro lado, houve uma regulação positiva de genes associados à adesão e às vias relacionadas à matriz extracelular (aumento do desenvolvimento da matriz extracelular), indicando restauração ativa da função muscular em casos de RE meses após o evento agudo.8
Em síntese, destacamos o que estas pesquisas nos ensinam:
Como o gene RYR1 afeta a função muscular
Associação de variantes (mutações) no RYR1 e Hipertermia Maligna (HM)
Como o defeito no RYR1 leva à Rabdomiólise
O que acontece durante a Rabdomiólise por Esforço (RE)
O que os indivíduos com variantes no RYR1 precisam saber
Hipótese
Referências Bibliográficas:
A Miopatia Congênita Centronuclear é uma doença que causa muitas exclamações, mas também, interrogações. Sempre que as pessoas chegam até mim para questionar sobre minha doença, elas fazem uma exclamação dizendo, “nunca tinha ouvido sobre sua doença !”, e em seguida questionam como ela me afeta, e mais uma vez elas exclamam dizendo, "puxa vida !", e por ultimo exclamam novamente, “essa é uma super rara !”, daí respondo, "sim, no pé da palavra, ela é uma Doença Rara ou talvez uma Doença Ultrarrara.
A Miopatia Congênita Centronuclear decorrente de mutações no gene RYR1 é classificada como Doenças Raras ou Doenças Ultrarraras ?
Entender essas condições é essencial para oferecer o suporte adequado e assegurar abordagens terapêuticas personalizadas tanto para o paciente quanto para seus familiares. Doenças raras nem sempre recebem a atenção que merecem porque afetam relativamente poucas pessoas. Muitas vezes, pode levar anos para que uma pessoa receba o diagnóstico correto de uma doença rara, e cerca de 95% das doenças raras e ultrarraras ainda não têm tratamento.
Diferentes partes do mundo, as doenças raras e ultrarraras são identificadas e tratadas de maneiras diferentes, mas de um modo genérico a definição é mais ou menos assim:
Uma Doença Rara é aquela que acomete no máximo 65 pessoas a cada 100.000 habitantes, o que corresponde a uma prevalência aproximada de 1 para cada 1.500 indivíduos. A maior parte dessas condições tem origem genética, embora algumas possam surgir devido a fatores infecciosos ou relacionados ao sistema imunológico.
A Doença Ultrarrara, como o próprio nome indica, corresponde a condições ainda mais incomuns, com uma incidência de cerca de 1 caso para cada 50.000 indivíduos. Por serem extremamente infrequentes e pouco conhecidas, essas doenças geralmente apresentam maior dificuldade diagnóstica e exigem cuidados altamente especializados e personalizados.
Com relação à questão levantada no início do texto, a prevalência exata da Miopatia Congênita Centronuclear causada especificamente pela mutação no gene RYR-1 é desconhecida.
A dificuldade em determinar um número exato ocorre porque:
• Ela é uma doença ultrarrara por estar dentro de um grupo de doenças já raras, as Miopatias Congênitas.
• Os dados epidemiológicos costumam ser apresentados para a Miopatia Congênita Centronuclear (MCCN) considerando o conjunto das patologias que compõem esse grupo, que inclui mutações nos genes MTM1, DNM2, BIN1 e RYR1.
• O gene RYR1 está associado a um espectro de doenças musculares conhecidas como Doenças Relacionadas ao RYR1 ou RYR1-DR, sendo a forma Centronuclear apenas uma das possíveis manifestações, juntamente com a Miopatia Central Core (MCC), e outras.
Embora não haja um número específico de prevalência para a Miopatia Congênita Centronuclear causada pela mutação no RYR1, podemos contextualizar com base nas informações disponíveis para as condições relacionadas:
• No grupo da Miopatia Congênita Centronuclear (MCCN) em geral, a prevalência geral do grupo é desconhecida. A forma mais comum dentro deste grupo é a Miopatia Miotubular (ligada ao X) causada por mutações no gene MTM1, cuja incidência é estimada em cerca de 1 em 50.000 indivíduos do sexo masculino.
• No grupo das Doenças Relacionadas ao RYR1 (RYR1-DR) em geral, a doença mais frequentemente associado é a Miopatia Central Core (MCC), que é a forma mais comum de miopatia congênita não distrófica.
Um estudo de uma série pediátrica na Espanha estimou a incidência de Miopatias Relacionadas ao RYR1, que abrange a Centronuclear e a Central Core, se mostra em cerca de 1 em 10.000 nascidos vivos na área de estudo.
Em síntese, a mutação no gene RYR1 está associada a um fenótipo de ocorrência rara. A prevalência específica do subtipo Centronuclear (MCCN) ainda não foi estabelecida de forma isolada na literatura médica, sendo considerada extremamente baixa. Entretanto, não há consenso atual sobre classificá-la como no grupo de Doenças Raras ou no grupo de Doenças Ultrarraras.
Na última postagem entitulada, A Importância das Intervenções No Estilo de Vida Para Controle Dos Sintomas da Miopatia Congênita Centronuclear, fiz uma abordagem sobre a importância das condutas multidisciplinar e individualizada, focada na reabilitação e no suporte contínuo para otimizar a funcionalidade e o bem-estar. Esse estilo de vida, seria sustentado por um tripé de condutas, sintetizado no Controle de Stress, Nutrição, e Atividade Física.
Seguindo essa linha de entendimento, gostaria de compartilhar com o leitor sobre minha experiência pessoal, enquanto portador de Miopatia Congênita Centronuclear causada pela mutação genética no RYR-1 com herança autossômica recessiva, com o acompanhamento nutricional que faço com a Dra Victoria Pitombo. Como tanto, segue as gentis palavras da Dra Victoria sobre nossa relação enquanto paciente, e logo a seguir você verá um estudo, base do trabalho aplicado no meu programa nutricional e de suplementação.
" Desde o início do nosso acompanhamento, o que mais me marca no Orlando é o comprometimento real que ele tem com a própria saúde. A nossa relação vai muito além de prescrições: é uma parceria construída com confiança, escuta e respeito por um corpo que exige atenção minuciosa devido à Miopatia Congênita Centronuclear. Cada ajuste que eu faço, desde a ingestão proteica até orientações de digestão e hidratação, tem um propósito maior: proteger sua musculatura, preservar funcionalidade, reduzir estresse oxidativo e garantir mais autonomia e qualidade de vida para ele.
Ao longo desses meses, construímos uma nutrição precisa e totalmente personalizada: calorias calibradas para preservar massa muscular e reduzir gordura abdominal, além de suplementos escolhidos para otimizar função neuromuscular sem gerar desconforto gastrointestinal. Tudo específico para o Orlando. Ver a evolução dele, melhora da autoestima, engajamento e confiança no processo (que ainda temos muito pela frente!) é uma das partes mais gratificantes do nosso trabalho juntos. " Dra Victoria Pitombo
NOTA IMPORTANTE: O estudo apresentado a seguir deve ser entendido como elemento de base científica, portanto, o programa de nutrição e suplementação para cada indivíduo deve ser altamente individualizado e baseado na avaliação clínica, estado nutricional atual e exames laboratoriais. Para garantir segurança e eficácia, as intervenções nutricionais e a suplementação devem ser sempre orientadas e conduzidas por um médico e um nutricionista especializados ou com conhecimento em doenças neuromusculares.
Dra. Victoria C. Pitombo - CRN 58214
℘ Nutricionista especializada a nível de Residência em doenças crônicas pelo Hospital do Coração (Hcor)
℘ Nutricionista Funciona(VP), Esportiva e com especialidade em Obesidade (USP)
Nutrição e Suplementação na Miopatia Congênita Centronuclear Associada à Mutação no Gene RYR1: Evidências Atuais e Considerações Clínicas
As miopatias relacionadas ao gene RYR1 englobam o grupo mais prevalente de miopatias congênitas, incluindo fenótipos como miopatia centronuclear. A mutação compromete a função do receptor rianodina, responsável pela liberação controlada de cálcio intracelular durante a contração do músculo. Como resultado, observam-se fraqueza de predomínio proximal, fadiga rápida, intolerância ao exercício, risco maior de hipertermia maligna e, em muitos casos, comprometimento respiratório.
Embora ainda não exista uma cura, percebe-se consenso crescente de que intervenções nutricionais e suplementação direcionada desempenham um papel importante no tratamento das miopatias. Isso inclui uma melhora na preservação da função muscular, na proteção óssea e na modulação do estresse oxidativo, todos aspectos centrais na fisiopatologia dessa condição.
1. Fundamentos do manejo nutricional
Adequação energética e composição corporal
Pacientes com miopatias congênitas apresentam gasto energético variável, podendo evoluir com desnutrição ou excesso de peso. Ambos são prejudiciais à função muscular e respiratória. Assim, a ingestão energética deve ser individualizada, sempre com foco na preservação da massa magra.
Proteína e estímulo anabólico
A literatura mostra que em doenças neuromusculares a ingestão proteica deve ser 1,2–1,5 g/kg/dia, distribuída ao longo do dia. Para alguns pacientes, pode-se extrapolar a ingestão para até 2g/kg/dia.
A distribuição da proteína ao longo do dia (em no mínimo 4 refeições), otimiza a síntese proteica muscular e reduz períodos de catabolismo, especialmente importante em indivíduos com fadiga crônica e menor resposta anabólica.
2. Suplementação baseada em plausibilidade fisiológica e evidência
Creatina Monohidratada
A creatina possui o potencial de aumentar a disponibilidade de fosfocreatina e melhora a ressíntese de ATP, mecanismo útil em condições que cursam com fadiga muscular rápida. Ensaios clínicos em outras doenças neuromusculares demonstram melhora modesta, mas consistente, na força e na resistência.
Dose usual: Em pacientes com Miopatia congênita centronuclear, o ideal é calcular a dose por kg de peso. Exemplo: Se o paciente pesa 67kg ele deve consumir 67kg x0,1 = 6.7g de creatina por dia.
Leucina
A leucina ativa a via mTOR, estimulando a síntese proteica muscular que é um processo frequentemente diminuído em miopatias com limitação funcional.
Na maioria das vezes, só o suporte de leucina via alimentos não é suficiente, então é interessante suplementar. Dose utilizada em estudos: 2–3 g junto com as refeições principais 2 a 3x por dia, dependendo do paciente. Ou, o paciente pode consumir a leucina 1x ao dia no período noturno.
N-acetilcisteína (NAC)
O estresse oxidativo é um dos mecanismos envolvidos na disfunção do RYR1. A NAC é precursora de glutationa e moduladora do sistema redox. Ensaios clínicos mostram segurança, embora os efeitos em marcadores oxidativos sejam variáveis.
Dose usual: 600–1200 mg/dia.
Outros antioxidantes relevantes
O desequilíbrio redox presente em mutações do RYR1 motiva o uso de antioxidantes adjuvantes:
Vitamina B12
A deficiência de B12 agrava fadiga, fraqueza e neuropatia, podendo intensificar limitações motoras já presentes.
Indicação: quando há deficiência documentada ou fatores de risco.
Doses usuais: 500–1000 mcg/dia (oral) ou protocolo IM médico.
Vitamina D e Cálcio - papel central para músculo e osso
Vitamina D
Há maior prevalência de deficiência de vitamina D em doenças neuromusculares, associada a redução da força, pior função respiratória e menor densidade óssea.
A suplementação e doses deve ser guiada por dosagem sérica (25-OH).
Cálcio
Por que o cálcio é tão importante nesses pacientes ?
O cálcio é o íon mais importante da contração muscular e justamente o elemento cuja movimentação é regulada através do receptor rianodina (RYR1). Em pacientes com mutações nesse gene, ocorrem algumas alterações no fluxo de Ca²⁺, como liberação excessiva ou dificuldade de recaptura, contribuindo para: fadiga precoce;maior risco de dano estrutural das fibras; dificuldade de relaxamento muscular e piora de sintomas ao esforço.
Além disso, muitos pacientes apresentam baixa densidade mineral óssea devido à imobilidade, baixa massa muscular, ingestão reduzida ou pouca exposição solar. Assim, o cálcio torna-se essencial tanto para a função muscular quanto para a prevenção de osteopenia e fraturas, que impactam diretamente mobilidade e autonomia.
Recomendações gerais:
L-Tirosina
A L-tirosina é precursora de catecolaminas e pode modular aspectos de vigília, foco e tolerância ao esforço, elementos frequentemente prejudicados em miopatias crônicas. Estudos sugerem benefício potencial em quadros de fadiga central.
Dose usada em protocolos clínicos: 500–2000 mg/dia.
Considerações finais
Embora a miopatia centronuclear por mutação no RYR1 não possua ainda um tratamento curativo, o manejo nutricional adequado e a suplementação estratégica apresentam papel importante no suporte à função muscular, no controle do estresse oxidativo, na proteção óssea e na melhoria da capacidade funcional diária.
Intervenções como adequação proteica, creatina, leucina, vitamina D e cálcio, NAC, antioxidantes, vitamina B12 e L-tirosina possuem fundamentos fisiológicos consistentes e vêm sendo incorporadas progressivamente à prática clínica, sempre de forma individualizada e integrada ao acompanhamento multiprofissional.
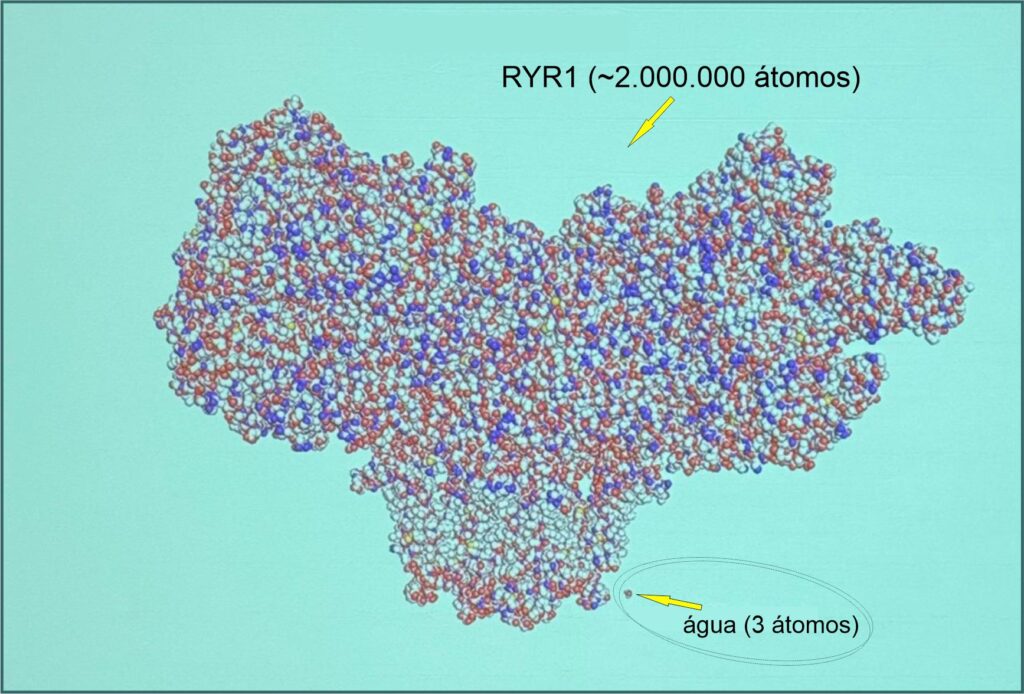
O RYR1 é um dos maiores genes do corpo humano, e é responsável para o funcionamento dos nossos músculos.
O gene RYR1 (Receptor de Rianodina 1) é um dos maiores e mais complexos genes do genoma humano. Ele contém mais de 100 exons e cerca de 15 mil pares de bases na sua sequencia de DNA, abrangendo uma grande extensão no cromossomo 19. A estimativa é que o gene RYR1 tenha cerca de 2 milhões de átomos, e esse é somente um cálculo de aproximação, pois a quantidade exata pode variar dependendo da sequência específica de nucleotídeos.
Para se ter uma idéia da complexidade do RYR1, ele é imensamente maior e mais complexo do que moléculas simples como da água, tão crucial para nossa vida, e a título de comparação, pasmem ! …o RYR1 tem 2 milhões de átomos, e a água, também conhecida como H20, tem somente 3 átomos.
O gene RYR1 que é crucial para a função do receptor de rianodina, que é um canal de cálcio localizado no Retículo 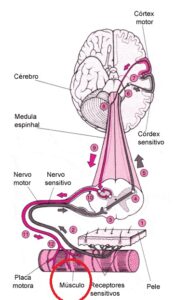 Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+
Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+) do RS para o citoplasma; 4). O cálcio dispara a contração muscular ao permitir a interação Actina-Miosina; 5). Para o relaxamento, o sinal cessa, o RYR1 se fecha, e rapidamente retorna o cálcio do citoplasma, devolvendo-o ao RS.
Em resumo, o RYR1 atua como o principal portão de saída do cálcio do reservatório intracelular. Ele é o mediador essencial que garante que o comando elétrico do nervo seja traduzido em movimento mecânico, controlando a entrada abrupta de cálcio que inicia a contração e, consequentemente, permitindo que a sua reabsorção finalize o processo para o relaxamento.
Como disse no início deste texto, devido ao tamanho e complexidade do RYR1, mutações nesse gene podem estar associadas a várias doenças musculares, incluindo:
O estudo científico do RYR1 é importante para entender essas condições ou doenças, e desenvolver tratamentos adequados em busca de minimizar seus efeitos e sintomas, pensando até na sua própria cura. Além disso, a análise genética pode ajudar em diagnósticos precoces e na prevenção de complicações associadas a essas doenças.
Durante o tempo que tenho acompanhado a movimentação de informações em torno das questões relacionadas ao RYR1, observo um grande esforço da comunidade científica em busca de entender o mecanismo de funcionamento deste complexo e grandioso gene, grande no tamanho, mas sobretudo grande em sua importância. Essas questões fazem com que o desafio ainda seja maior não só por conta dos indivíduos que têm uma doença a ele relacionada, mas também para compreensão sobre o impacto do envelhecimento celular em qualquer outro indivíduo. Observo também um grande movimento de trabalhos sendo desenvolvidos por cientistas de toda parte do mundo em busca de desenvolver uma droga para tratamentos adequados, para de repente ao menos minimizar seus efeitos e sintomas, até sua cura das doenças relacionadas ao RYR1. Particularmente, minha percepção é que as pesquisas através da técnica de reposicionamento de drogas, também chamado de redirecionamento de fármacos têm se mostrado mais promissoras no curto prazo. O “reposicionamento de drogas” se trata de uma estratégia que busca descobrir novas aplicações terapêuticas para medicamentos já existentes, que originalmente foram desenvolvidos para tratar outras doenças. Essa técnica tem a vantagem por seus reduzidos custos, assim como pela redução de tempo gasto no processo de desenvolvimento, e menores riscos no uso da eventual droga, pois já se conhece a farmacocinética e toxicidade do medicamento pesquisado, um exemplo real e prático de pesquisa em curso está se dando com o Sulfato de Salbutamol.
Contudo, a terapia genética também tem se mostrado uma abordagem promissora para o tratamento e cura de diversas doenças genéticas, oferecendo a possibilidade de corrigir ou substituir genes defeituosos. Embora este tipo de tratamento ainda esteja em desenvolvimento e não seja uma solução universal para algumas doenças, como por exemplo certas formas de miopatias ou doenças hereditárias, os resultados das pesquisas têm sido encorajadores. No entanto, a eficácia e a segurança da terapia genética podem variar dependendo da doença, do tipo de terapia utilizada, e condições do paciente. Além disso, questões éticas e de custo também são consideradas importantes. Contudo, enquanto a terapia genética oferece uma expectativa otimista, é uma área em evolução que requer mais pesquisas, altos recursos financeiros, e testes clínicos para se consolidar como a melhor solução para todas as doenças genéticas.
No caso específico das doenças relacionadas à mutação do gene RYR1, a terapia genética apresenta vários desafios, principalmente, como escrito no início deste texto, devido ao tamanho do gene, complexidade das doenças relacionadas a ele, mas também por considerar que os músculos representam aproximadamente 40% da massa total do corpo humano, e a maior parte dessa massa muscular é composta por músculos esqueléticos. Observe a seguir algumas das dificuldades:
Esses desafios tornam a pesquisa e o desenvolvimento de terapias para mutações no gene RYR1 complexos e exigem abordagens inovadoras e multidisciplinares.
Eu, enquanto portador de uma doença causada pela mutação nesse “grandioso” RYR1, tenho uma relação muito particular com esse gene, e a cada dia que passa o conheço um pouco mais, ..... confira minha próxima postagem intitulada “Eu vi o meu gene em 3D e entendi o que acontece dentro de mim”.
O SORRYR-1 mais uma vez cumpriu com seu objetivo de informar sobre as doenças relacionadas ao RYR-1 aos indivíduos afetados pela doença e seus familiares, profissionais da área da saúde, além de apoiar e participar de pesquisas científicas. Assim como já foi relatado nas redes sociais, eu fui convidado pela Dra Lizan Stinissen, do departamento de neurologia da Radboud University Medical Center, de Nijmegen, Holanda, para colaborar na elaboração de uma pesquisa internacional dirigida a pacientes com doenças relacionadas ao RYR1 visando sua participação em ensaios clínicos. O trabalho de elaboração da pesquisa foi concluído, pesquisa aplicada, e seu resultado foi apresentado pela Dra Lizan durante a Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos, tendo no final da apresentação, o orgulho de ouvir palavras de reconhecimento pela nossa participação e contribuição. O estudo se concentrou no que as pessoas com Miopatias Congênitas RYR1 e seus familiares esperam de futuros ensaios clínicos, e o que os incentivaria a participar dessas pesquisas. O objetivo deste estudo é fornecer aos pesquisadores clínicos, às empresas farmacêuticas, e à comunidade de indivíduos afetados pelas doenças relacionadas ao RYR-1, subsídios ao desenvolvimento de pesquisa em ensaios clínicos que sejam significativas e de importância terapêutica para os pacientes. O resultado da pesquisa fornecerá também aos profissionais de saúde uma visão sobre os sintomas mais importantes para os pacientes e as lacunas nas terapias disponíveis para os indivíduos com miopatia relacionada ao RYR1.
Medical Center, de Nijmegen, Holanda, para colaborar na elaboração de uma pesquisa internacional dirigida a pacientes com doenças relacionadas ao RYR1 visando sua participação em ensaios clínicos. O trabalho de elaboração da pesquisa foi concluído, pesquisa aplicada, e seu resultado foi apresentado pela Dra Lizan durante a Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos, tendo no final da apresentação, o orgulho de ouvir palavras de reconhecimento pela nossa participação e contribuição. O estudo se concentrou no que as pessoas com Miopatias Congênitas RYR1 e seus familiares esperam de futuros ensaios clínicos, e o que os incentivaria a participar dessas pesquisas. O objetivo deste estudo é fornecer aos pesquisadores clínicos, às empresas farmacêuticas, e à comunidade de indivíduos afetados pelas doenças relacionadas ao RYR-1, subsídios ao desenvolvimento de pesquisa em ensaios clínicos que sejam significativas e de importância terapêutica para os pacientes. O resultado da pesquisa fornecerá também aos profissionais de saúde uma visão sobre os sintomas mais importantes para os pacientes e as lacunas nas terapias disponíveis para os indivíduos com miopatia relacionada ao RYR1.
Veja a seguir como tudo se deu...
Pesquisa com Paciente com Doença Relacionada ao RYR1
Este estudo se concentra sobre as preferências de ensaios clínicos de indivíduos com doenças relacionadas ao RYR1 (RYR1-RD).
Antecedentes e Objetivo do Estudo
Atualmente, existem vários estudos pré-clínicos e estudos de história natural em andamento sobre o RYR1-RD, assim como já houve dois ensaios clínicos até agora. Os possíveis próximos ensaios clínicos que estão por vir indicam ter bons resultados. A comunidade de pacientes pode contribuir para esses futuros ensaios apontando suas preferências e expectativas. Espera-se que este estudo possa fornecer aos pesquisadores clínicos, empresas farmacêuticas, e à comunidade de pacientes informações de importância significativas para pesquisas terapêuticas aos pacientes. O estudo destaca os maiores sintomas e queixas, buscando identificar medidas de relevantes resultado.
Este estudo é uma continuação de uma publicação sobre uma pesquisa online com pacientes iniciada pela Fundação RYR-1, combinada com depoimentos de pacientes apresentados durante o Workshop Internacional de Pesquisa realizado em 2022. O relatório completo do estudo anterior intitulada como: “Indivíduos e Famílias Afetados por Doenças Relacionadas ao RYR1: A Perspectiva do Paciente/Cuidador”, pode ser encontrado disponível no seguinte link aqui: https://ryr1.org/medical-literature/individuals-and-families-affected-by-ryr1-related-diseases-the-patient-caregiver-perspective
Como o Estudo Foi Estruturado
A pesquisa com pacientes RYR1-RD esteve disponível online entre 10 de março e 5 de maio de 2025. Ela incluiu perguntas sobre a condição dos participantes e como ela estava sendo tratada na época, além de suas opiniões e expectativas em relação às pesquisas e ensaios clínicos. A pesquisa foi disponibilizada em 6 idiomas: inglês, espanhol, português, francês, alemão e holandês. Como apoio ao processo de tradução e revisão da pesquisa, contamos com a colaboração de perto de membros da Fundação RYR-1 e palestrantes de acordo com o seu respectivo idioma, muitos dos quais eram também pacientes.
Primeiros Resultados e Próximos Passos
Os resultados fornecem informações sobre:
Um total de 152 pessoas, representando 19 países diferentes, responderam a pesquisa. Os resultados foram apresentados na Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos. No momento, estamos escrevendo um artigo científico com base nos resultados da pesquisa. Planejamos submetê-lo a uma revista médico-científico, para que tanto médicos quanto pesquisadores possam se beneficiar dos resultados. Esperamos que os resultados aumentem a compreensão dos pacientes sobre sua participação futura em pesquisas clínicas, assim como apoie os pesquisadores no desenvolvimento futuros de seus projetos de pesquisas clínicas. O artigo demonstrará a importância de refletir sobre as experiências, pensamentos e expectativas dos pacientes, e destaca a realização de um trabalho de cocriação com pesquisadores acadêmicos e a comunidade de pacientes. Uma atualização seguirá assim que o artigo for finalizado.
em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos. No momento, estamos escrevendo um artigo científico com base nos resultados da pesquisa. Planejamos submetê-lo a uma revista médico-científico, para que tanto médicos quanto pesquisadores possam se beneficiar dos resultados. Esperamos que os resultados aumentem a compreensão dos pacientes sobre sua participação futura em pesquisas clínicas, assim como apoie os pesquisadores no desenvolvimento futuros de seus projetos de pesquisas clínicas. O artigo demonstrará a importância de refletir sobre as experiências, pensamentos e expectativas dos pacientes, e destaca a realização de um trabalho de cocriação com pesquisadores acadêmicos e a comunidade de pacientes. Uma atualização seguirá assim que o artigo for finalizado.
RYR1-RD patient survey_LizanStinissen






Em julho de 2022, a Fundação RYR1 promoveu o Workshop Internacional de Pesquisa sobre Doenças Relacionadas ao RYR-1: Dos Mecanismos aos Tratamentos, foi o primeiro workshop internacional de pesquisa dedicado exclusivamente às doenças relacionadas ao RYR1 . O evento reuniu 45 pesquisadores presenciais e 10 virtuais de 11 países diferentes, todos envolvidos em pesquisas na busca de tratamento das doenças relacionadas ao RYR1, RYR1-RD (veja a galeria de imagens do evento). Sob a coordenação da co-diretora Brentney Simon, fui convidado, juntamente com um time de 10 indivíduos afetados por RYR1-RD para compartilhar nossa vivência, conhecimentos e interagir com os presentes. O objetivo científico do workshop foi promover um fórum que unisse os principais especialistas internacionais em doenças RYR1 (pesquisadores, clínicos e geneticistas) com indivíduos afetados, familiares e defensores de pacientes para compartilhar conhecimento, trocar ideias, formar colaborações e desenvolver novas estratégias para encontrar terapias eficazes.
Além do mais, nosso time desenvolveu e conduziu uma pesquisa com aplicação on-line, dirigida a 227 indivíduos, permitindo que pessoas afetadas por RYR1-RD tivessem suas vozes ouvidas através dos dados coletados. Nossos esforços foram tão bem-sucedidos que a Dra. Nicol C Voermans (Department of Neurology, Radboud University Medical Center, The Netherlands - click aqui e conheça seu histórico), uma das cientistas presentes, convidou Brentney para colaborar com sua equipe para compartilhar esses insights com profissionais médicos, além da comunidade RYR-1, na forma de um Artigo de Pesquisa.
Estou muito orgulhoso de ter feito parte deste importante projeto, dado sua importância médico-científica, à medida que o mesmo poderá contribuir nos avanços em busca de tratamento para as doenças relacionadas ao RYR1 (RYR1-RD).
Confira a seguir a publicação original (click no link ) ⇒ https://pubmed.ncbi.nlm.nih.gov/39150833
....ou a publicação original em pdf ⇒ https://content.iospress.com/download/journal-of-neuromuscular-diseases/jnd240029?id=journal-of-neuromuscular-diseases%2Fjnd240029
.... ou confira a seguir nesta postagem, a tradução e adaptação para a lingua portuguesa da publicação entitulada "INDIVÍDUOS E FAMÍLIAS AFETADAS POR DOENÇAS RELACIONADAS AO RYR1 - A PERSPECTIVA DO PACIENTE/CUIDADOR".
⇓ ⇓ ⇓ ⇓
Publicação em 13 Julho de 2024
Resumo Histórico e objetivo: Variantes patogênicas do RYR1, o gene que codifica o principal canal de liberação de cálcio do retículo sarcoplasmático (RyR1) com um papel crucial no acoplamento excitação-contração da célula muscular, estão entre as causas genéticas mais comuns de distúrbios neuromusculares não distróficos. Recentemente, conduzimos um estudo via questionário com foco em deficiências funcionais, fadiga e qualidade de vida (QV) em pacientes com doenças relacionadas ao RYR1 (RYR1-RD) em todo o espectro de doenças reconhecidas. Neste referido estudo via questionário, a perspectiva médica foi adotada, refletindo um protocolo de estudo projetado por neurologistas e psicólogos. Com este estudo em questão, queríamos abordar especificamente a perspectiva do paciente.INTRODUÇÃO
Variantes patogênicas do RYR1, o gene que codifica o principal canal de liberação de cálcio do retículo sarcoplasmático (SR) (RyR1) com um papel crucial no acoplamento excitação-contração (ECC) nas célular musculares, são uma das causas genéticas mais comuns de distúrbios neuromusculares não distróficos. As variantes patogênicas do RYR1 dão origem a uma ampla variedade de doenças relacionadas ao RYR1 (RYR1-RD) que se apresentam ao longo da vida, variando de miopatias congênitas de início precoce a manifestações episódicas na idade adulta, incluindo hipertermia maligna durante anestesia e rabdomiólise por esforço em indivíduos que de outra forma seriam amplamente saudáveis [1, 2]. Esse amplo espectro clínico associado ao RYR1 é devido ao impacto funcional altamente variável de diferentes variantes patogênicas do RYR1, modo de herança, bem como tipo e localização de mutações específicas.
Recentemente, conduzimos um estudo via questionário com foco em deficiências funcionais, fadiga e qualidade de vida (QV) em pacientes holandeses exibindo todo o espectro clínico de RYR1-RD [3]. Pudemos demonstrar que tanto o RYR1-RD permanente quanto o episódico estão associados a um impacto substancial da doença, caracterizado por limitações funcionais e fadiga grave, resultando em perda significativa da QV. Os resultados deste estudo aumentaram a conscientização e o reconhecimento dos sintomas típicos em indivíduos afetados por RYR1-RD. Além disso, o estudo também melhorou o gerenciamento do paciente e definiu áreas específicas que precisam de mais pesquisas, particularmente sobre o impacto da doença associado a esses distúrbios neuromusculares comuns.
A perspectiva adotada no estudo de questionário anterior foi a perspectiva médica distinta, refletindo um protocolo de estudo elaborado por neurologistas e psicólogos. Muitos clínicos, no entanto, permanecem em grande parte inconscientes do impacto do RYR1-RD na vida diária de seus pacientes e de suas expectativas em relação a tratamentos futuros. Esses aspectos estão gradualmente se tornando um tópico de pesquisa em outras doenças neuromusculares [4, 5], mas ainda não foram abordados no RYR1-RD. Para abordar essas questões, os membros da RYR-1 Foundation, uma associação internacional de pacientes dedicada ao RYR1-RD, iniciaram uma pesquisa online antes de um workshop científico e da conferência internacional de pacientes dedicado a essas condições. Os resultados desta pesquisa demonstram a forte necessidade do paciente de compartilhar suas perspectivas em primeira mão com os principais especialistas internacionais em RYR1.
O “RYR-1-Related Diseases International Research Workshop: From Mechanisms to Treatments”, realizado de 21 a 22 de julho de 2022 em Pittsburg (PA, EUA), foi o primeiro workshop internacional de pesquisa liderado por pacientes dedicado exclusivamente ao RYR1-RD [6]. O objetivo científico do workshop era fornecer um fórum que conectasse os principais especialistas internacionais em doenças RYR1 (pesquisadores, clínicos e geneticistas) com indivíduos afetados, familiares e defensores de pacientes para compartilhar conhecimento, trocar ideias, formar colaborações e desenvolver novas estratégias para encontrar terapias eficazes. Objetivos adicionais eram desenvolver recomendações de consenso para prioridades clínicas/de pesquisa, identificar itens acionáveis necessários para levar o campo adiante e fornecer uma plataforma para os estagiários se envolverem com pacientes/familiares de RYR1 e líderes estabelecidos no campo. Durante este workshop, a equipe do estudo apresentou os resultados da pesquisa on-line e convidou doze pacientes a apresentarem seus depoimentos sobre viver com RYR1. Este manuscrito descreve os resultados da pesquisa do paciente e dos depoimentos individuais, usando uma abordagem de métodos mistos; empregando uma análise quantitativa da pesquisa e uma análise qualitativa dos depoimentos.
MÉTODOS
Equipe de estudo
A equipe de estudo que projetou e executou a pesquisa consistiu em 14 indivíduos, incluindo indivíduos afetados, familiares e defensores preocupados com RYR1-RD. Três desses indivíduos (DH, BS, JR) participaram de reuniões virtuais de especialistas científicos e médicos (AS, RD, NV) envolvidos no planejamento e organização do workshop internacional (de novembro de 2021 a junho de 2022).
Plano da pesquisa on-line
A equipe do estudo discutiu uma série de tópicos, com base no que ouviram de diferentes pacientes em discussões internas na RYR-1 Foundation, para gerar o primeiro conjunto de perguntas para avaliar e entender os desafios diários enfrentados por indivíduos que vivem com RYR1-RD. O conjunto resultante de perguntas foi posteriormente discutido com os especialistas científicos e médicos para avaliar as perguntas mais relevantes e chegar a um consenso sobre as perguntas finais com base na importância e relevância clínica. Isso resultou em uma pesquisa de 21 perguntas cobrindo cinco temas diferentes: Demografia (1–3); Diagnóstico (4–9); Sintomas e impacto da condição (10–12); Atividade física (13–16); e Pesquisa e estudos clínicos (17–21). A pesquisa completa está incluída como Dados suplementares A: Pesquisa on-line.
Esta pesquisa usou um design transversal descritivo e estava disponível para todos os indivíduos com RYR1-RD que atendiam aos critérios de inclusão por meio da plataforma de pesquisa on-line 'SurveyMonkey'. A pesquisa esteve ativa de 9 de maio a 25 de junho de 2022. Os critérios de inclusão incluíram: 1) Ter recebido um diagnóstico médico de RYR1-RD; e 2) Ser capaz de entender e escrever em inglês. Para indivíduos <5 anos de idade, os pais ou responsáveis legais foram convidados a responder à pesquisa. Indivíduos entre 5 e 17 anos foram convidados a envolver seus pais ou responsáveis legais para responder às perguntas.
Uma abordagem de amostragem proposital foi usada, e os quadros de amostragem incluíram: páginas do Facebook (FB), Twitter e Instagram da "The RYR-1 Foundation", "ryr1.org"; grupo do FB "RYR-1 Families"; página do FB "Living with RYR-1- Support Group"; página do FB "Central Core Disease & Minicore: a place for support, learning & friends" e lista de e-mail da The RYR-1 Foundation. As instruções específicas fornecidas foram: "Forneça o melhor palpite se você não tiver certeza absoluta"; e ‘Pule qualquer pergunta que você não queira responder’. Os participantes foram informados de que o objetivo da pesquisa era ajudar pesquisadores e especialistas a aprender mais e entender melhor o impacto de viver com um RYR1-RD, e ajudar pesquisadores a desenvolver novas estratégias para identificar e avaliar terapias adequadas.
Convite e instruções para depoimentos
Primeiramente, os pacientes que são membros da RYR1 Foundation foram convidados por e-mail. Daqueles que relataram estar interessados e da equipe do estudo, os participantes foram selecionados com base na idade, sexo, raça e tipo e gravidade do diagnóstico para representar uma ampla gama de indivíduos afetados por RYR1-RD. Essa seleção foi feita pelos pacientes que participaram do grupo de estudo. Um grupo de 12 indivíduos que foram direta ou indiretamente afetados por um RYR1-RD foram convidados a fornecer breves depoimentos (cerca de 7 minutos) durante o workshop internacional que resumem sua perspectiva sobre como é viver ou cuidar de alguém com RYR1-RD. Esses indivíduos foram instruídos a falar sobre o processo de diagnóstico, sintomas e impacto do RYR1-RD em sua vida diária, bem-estar físico e mental, seus tratamentos passados e atuais e expectativas de pesquisas futuras. Os participantes foram encorajados a incluir experiências, preocupações e ideias específicas para pesquisadores e especialistas médicos. As instruções escritas para os depoimentos são adicionadas como Dados suplementares B: Instruções para depoimentos.
Considerações éticas
As informações foram coletadas sem pedir aos entrevistados que revelassem suas identidades. Consequentemente, as informações enviadas não foram consideradas informações de saúde protegidas. Portanto, o Health Insurance Portability and Accountability Act (WMO)* não precisou controlar o uso dessas informações. Apesar disso, a RYR-1 Foundation manteve salvaguardas rígidas para proteger as informações do paciente de uso ou divulgação que não sejam consistentes com seus objetivos. Além disso, os autores realizaram aquisição, análise, interpretação e publicação de dados de acordo com as diretrizes de boas práticas clínicas da Radboud University Medical Center. Finalmente, o Radboud Research Ethics Committee considerou esta pesquisa não sujeita ao WMO (2024-17388).
Armazenamento e análise de dados
Dados quantitativos. Os dados da pesquisa foram armazenados anonimamente em um arquivo Excel e, posteriormente, transferidos para o SPSS. Estatísticas descritivas foram predominantemente usadas para caracterizar os dados (SPSS versão 27, IBM, Armonk, Nova York). Os gráficos foram preparados no software Graphpad Prism versão 9.5.0 (Graphpad Software, San Diego, CA, EUA). Os testes rho de Spearman foram usados para avaliar a relação entre sintomas, entre estado de deambulação e sintomas, idade do diagnóstico e sintomas, e estado de deambulação e frequência de exercícios. Os participantes também foram divididos em duas faixas etárias para comparar crianças a adultos (idade até 17 e ≥18 anos). Uma correção de Bonferroni foi aplicada para neutralizar múltiplas comparações entre os diferentes grupos de doenças RYR1-RD. ANOVAs unidirecionais foram usadas para comparar os diferentes RYR1-RD com relação ao número e tipo de sintomas físicos e psicológicos.
Dados qualitativos. Os pacientes que apresentaram seus depoimentos foram pseudoanonimizados: nas transcrições e análises, apenas os números dos participantes foram usados. Coletamos dados demográficos dos participantes, incluindo idade, sexo, diagnóstico e papel na comunidade de pacientes (paciente/pai/outro cuidador). Todos os depoimentos foram gravados em áudio. Os depoimentos que foram apresentados apenas verbalmente (n=9/12) foram transcritos na íntegra. Todos os depoimentos escritos foram complementados com informações adicionais das gravações. Os depoimentos escritos e transcritos na íntegra foram carregados no software Atlas-ti versão 8.1 e analisados usando uma análise temática [7, 8]. A análise temática é um método para identificar, analisar e relatar temas dentro dos dados [9]. Dois pesquisadores (LS e NV) analisaram os dados de forma independente por meio de um processo de comparação e raciocínio indutivos, partindo dos dados e do objetivo e não de teorias preexistentes. Esse objetivo era obter uma melhor compreensão da vida de um paciente afetado por um RYR1-RD, e expectativas e desejos em relação ao futuro. Os dois pesquisadores buscaram independentemente as unidades básicas de significado segmentando os dados e dando a esses segmentos rótulos conceituais que estavam intimamente relacionados às palavras dos participantes (codificação aberta). Ambos os autores compararam e discutiram esses códigos até chegarem a um consenso. Em seguida, eles identificaram relações entre os códigos abertos e agruparam esses códigos referentes ao mesmo fenômeno em categorias (codificação axial).
As categorias estavam parcialmente sobrepostas aos temas da pesquisa: Diagnóstico; Sintomas e impacto da condição; Tratamento e Expectativas. Para os temas principais, subtemas foram criados (vida antes do diagnóstico, diagnóstico, sintomas, efeito no funcionamento físico, efeito na vida diária, efeito na saúde mental, tratamento, expectativas e obtenção de conhecimento) e com três pesquisadores (LS, SC, NV) as citações mais significativas foram selecionadas por subtema que melhor representava os temas.
RESULTADOS
Um resumo conciso dos resultados da pesquisa e dos depoimentos foram incluídos no relatório do workshop [6]. Apresentamos aqui o conjunto de dados completo, começando com os dados quantitativos seguidos por dados qualitativos sobre os seguintes temas: 1) Diagnóstico; 2) Sintomas e impacto da condição; 3) Atividade física; 4) Tratamento; 5) Pesquisa e estudos clínicos e 6) Expectativas. Os temas 1 e 2 foram rediscutidos tanto na pesquisa quanto no depoimento. Os temas 3 e 5 foram discutidos apenas na pesquisa e os temas 4 e 6 apenas nos depoimentos. Nos depoimentos, todos os temas foram divididos em diferentes aspectos-chave.
RESPOSTAS
Pesquisa - A pesquisa foi respondida por 227 pacientes, pais ou outros cuidadores (143 mulheres e 84 homens). Para as perguntas que não foram respondidas por todos os participantes, é indicado quantos pacientes responderam às perguntas. As idades dos participantes variaram de 1 a 85 anos, com média de 37 ± 21 anos.
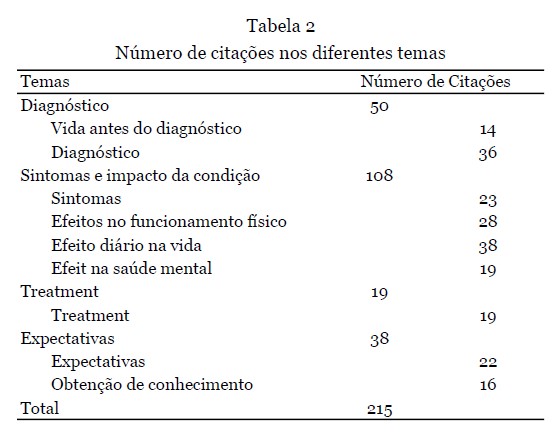
Depoimentos - 12 depoimentos foram apresentados durante o workshop. Nove deles foram fornecidos como texto escrito e complementados com a gravação de áudio, e os outros três estavam disponíveis apenas como gravação de áudio e transcritos na íntegra. Os dados demográficos dos participantes que forneceram os depoimentos são apresentados na Tabela 1. A análise qualitativa resultou na seleção de 215 citações, refletindo os quatro temas principais (Tabela 2).
Abaixo, descrevemos os resultados da pesquisa seguidos pelos resultados dos depoimentos de acordo com os principais temas de cada ferramenta de coleta de dados, conforme explicado na seção de métodos. Números de pacientes e porcentagens são relatados na pesquisa. Os casos em que nem todos os participantes responderam a uma pergunta específica são explicitamente indicados.
DIAGNÓSTICO
Resultados da Pesquisa - A seção de diagnóstico da pesquisa consistiu em seis perguntas. A idade dos entrevistados no diagnóstico foi variável, variando de 1 a > 60 anos. 22% dos participantes declararam que foram diagnosticados com RYR1-RD antes dos 5 anos de idade (Fig. 1). Os pacientes foram diagnosticados apenas por testes genéticos (n = 99/227,44%), por biópsia muscular e testes genéticos (n = 85/227,37%), ou apenas por biópsia muscular (n = 28/227,12%). Em um pequeno subconjunto de casos, o diagnóstico foi presumido com base no fenótipo e na presença de um parente de primeiro grau afetado com diagnóstico confirmado de RYR1-RD (n = 6/227,3%). Oito pacientes (4%) indicaram outras combinações dos modos de diagnóstico acima mencionados e um paciente não indicou o modo de diagnóstico.
Houve uma ampla gama de RYR1-RD, com a Miopatia Central Core (CCD) sendo a mais comum (n = 92/173, 53%). Suscetibilidade à Hipertermia Maligna (MHS) (n = 29/173,17%), Miopatia Centronuclear (CNM) (n = 10/173,6%), Desproporção Congênita do Tipo de Fibra (CFTD) (n = 9/173,5%) e Doença Multi-minicore (MmD) (n = 6/173,3%) foram outros diagnósticos, mas menos frequentes. 18 participantes indicaram "outro" para esta questão, com explicações de "incerto", "não especificado" ou "nova variante" (Fig. 2A). Nove pacientes tiveram um diagnóstico de MHS e miopatia. A herança foi autossômica dominante (AD) (n=61/215,28%), autossômica recessiva (AR) (n=59/215,27%), ou de novo ou espontânea (n=21/215,10%). Muitos dos participantes relataram não saber o modo de herança (n=74/215;34%) (Fig.2B).
Os participantes relataram sua capacidade física atual como capaz de andar sem assistência (n=145/223,65%), capaz de andar com assistência (n=27/223,12%), requer assistência de cadeira de rodas (n=13/223,6%) e requer uso de cadeira de rodas em tempo integral (n=38/223,17%). Pacientes com uma mutação ARordenovo necessitaram mais frequentemente do uso de cadeira de rodas em tempo integral (30,5% e 38% respectivamente, vs 5%, p < 0,001). Quase metade dos participantes considerou seus sintomas como progressivos (n = 107/223,47%), e um terço os relatou como estáveis (n = 77/223,34%). Outros não foram capazes de avaliar sua progressão devido a comorbidades ou sintomas variáveis.
Resultados dos depoimentos - Um total de 50 citações ilustraram o processo de diagnóstico (Tabela 3). Vários indivíduos mencionaram uma longa jornada de diagnóstico de meses a vários anos, chegando à idade adulta (início) quando foram diagnosticados, embora o início da doença tenha sido no nascimento. Alguns desses pacientes receberam um diagnóstico inicial incorreto, incluindo doença mitocondrial ou paralisia cerebral, antes que o diagnóstico correto de um RYR1-RD fosse finalmente estabelecido.
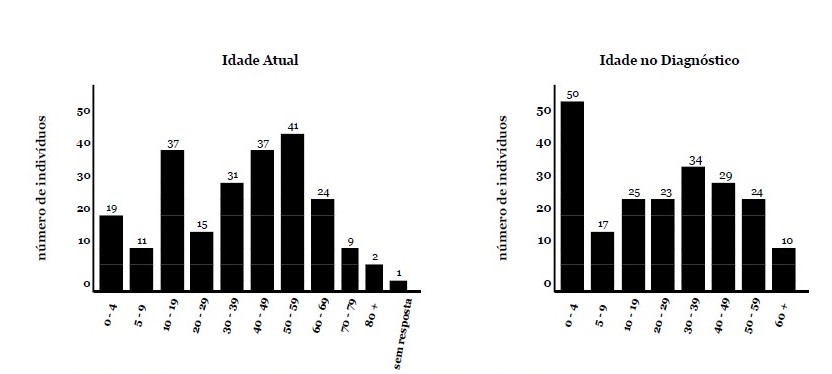
Fig. 1. A idade atual e a idade no diagnóstico. O eixo y descreve o número de pacientes para cada faixa etária. Esses histogramas mostram a ampla faixa da idade atual dos participantes e a idade no diagnóstico.
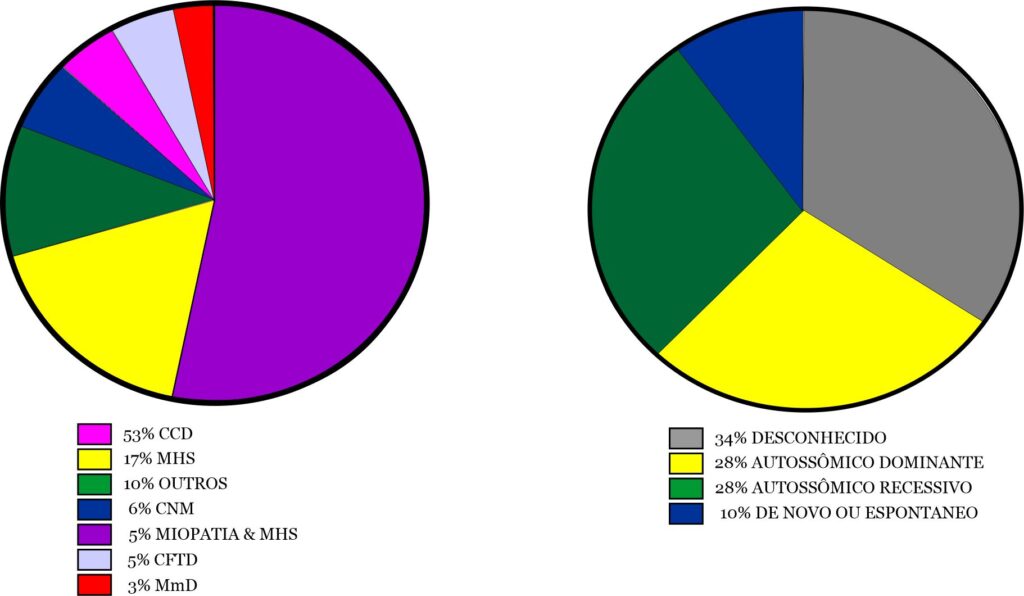
Fig. 2A. Genótipos dos pacientes incluídos. CCD: miopatia central core; MHS: suscetibilidade à hipertermia maligna; Outro: variante incerta, não especificada ou nova; CNM: miopatia centronuclear; CFTD: miopatia de desproporção do tipo de fibra congênita; MmD: miopatia multi-minicore. N=173. - Fig. 2B. Os diferentes modos de herança de RYR1-RD (N=215).
A vida antes do diagnóstico - Nas décadas de 1980 e 1990, pouca informação era conhecida sobre RYR1-RDs. O padrão de fraqueza, idade de início, sintomas específicos e a gravidade dos sintomas frequentemente resultavam em um amplo diagnóstico diferencial, e biópsias musculares eram frequentemente necessárias para confirmar o diagnóstico. Em geral, os pacientes comentaram sobre a dificuldade de ser diagnosticado com uma doença rara. Os pacientes ressaltaram o início precoce dos sintomas, incluindo atraso no desenvolvimento, mas também como esses primeiros sinais eram frequentemente ignorados ou reconhecidos somente após vários anos.
Diagnóstico - Em geral, os pacientes mencionaram seu histórico familiar de RYR1-RDs por meio de seus pais, irmãos ou outros membros da família, remontando a gerações. Eles relataram uma variabilidade intrafamiliar da gravidade da doença e a maneira como vivenciaram seu próprio diagnóstico ou o diagnóstico de seu filho. Um paciente mencionou que teve que esperar meses para consultar um especialista neuromuscular e, para alguns pacientes, levou anos para serem diagnosticados. Isso causou muito estresse para o paciente e sua família, principalmente por causa da incerteza associada. Pais sem formação médica enfrentaram desafios significativos para encontrar o caminho correto para o diagnóstico. Outros pais mencionaram que se sentiam solitários, pois não conheciam mais ninguém com uma experiência semelhante. Além disso, esses pais expressaram como a Fundação RYR-1 e a comunidade de pacientes os ajudaram. No geral, os pacientes descreveram seu processo de diagnóstico como uma longa jornada com muitas visitas ao hospital, testes adicionais e muitas perguntas sem resposta. Um dos pais mencionou o alívio que sentiu quando finalmente teve uma resposta para o diagnóstico de seu filho, embora não fosse o diagnóstico que esperavam ouvir. A maioria dos pais também expressou suas preocupações e estresse sobre a vida futura após o diagnóstico.

SINTOMAS E IMPACTO DA CONDIÇÃO
Resultados da pesquisa - Esta parte da pesquisa consistiu em três perguntas. A primeira pergunta sobre sintomas gerais propôs várias opções de resposta, incluindo, entre outras, fraqueza muscular geral, fadiga e dificuldades respiratórias, com uma opção aberta para listar outros sintomas. Os pacientes foram convidados a relatar todos os sintomas que se aplicavam a eles. Fraqueza muscular geral foi a mais frequentemente relatada (n = 190/227, 84%), seguida por fraqueza nas pernas (n = 185/227, 82%), dificuldade para correr (n = 183/227, 81%), dificuldade com escadas e para se levantar (n = 177/227, 78%) e fadiga (n = 168/227, 74%) (Fig. 3A e B). Para os sintomas acima mencionados, o grupo MHS teve uma frequência significativamente menor, exceto para fadiga, em comparação ao grupo miopatia. Não foram encontradas diferenças para esses sintomas entre diferentes faixas etárias (dados não mostrados). Também não houve correlação entre a idade do diagnóstico e o número de sintomas (r(210)=–0,79, p=0,253).
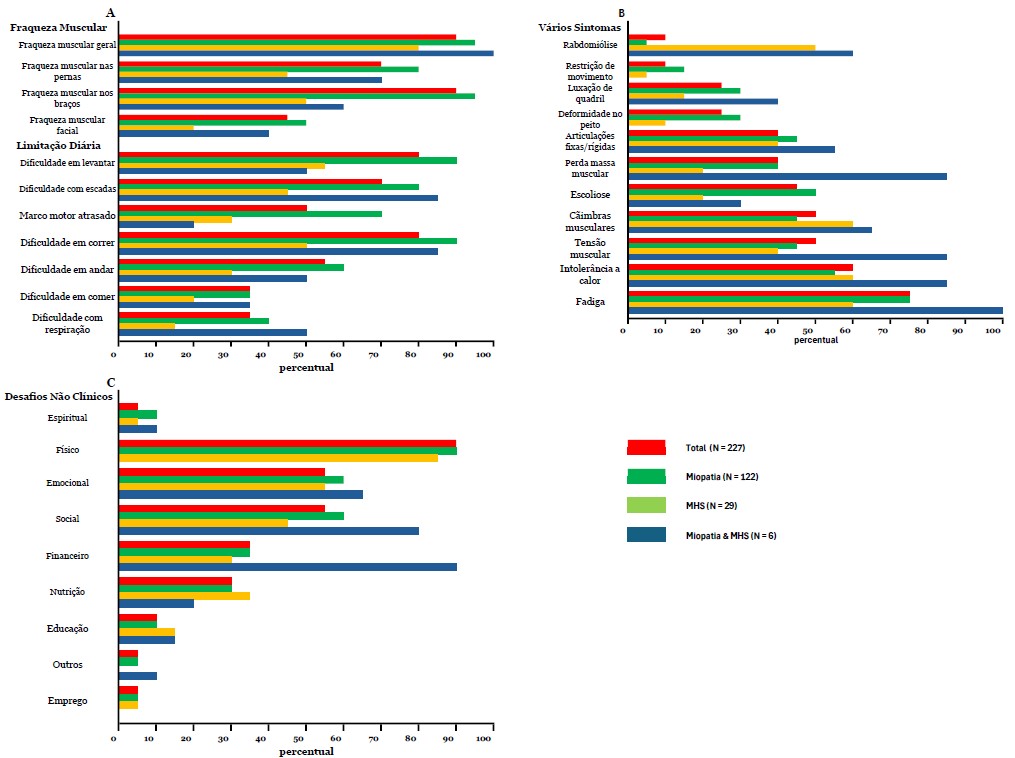
Fig. 3A, 3B, 3C. Sintomas Relatados - A porcentagem de pacientes por grupo RYR1-RD divididos em A) Fraqueza muscular e limitações na vida diária, B) Vários sintomas e C) Desafios não clínicos.
Os pacientes que necessitaram de uso integral de cadeira de rodas (n=38) ou caminharam com assistência (n=13) apresentaram significativamente mais sintomas do que aqueles capazes de caminhar sem assistência (n=145) (número médio de sintomas 13,5±3,3 e 11,6±3,8 vs. 9,12±4,2 respectivamente; p<0,001). Aqueles que necessitaram de uso integral de cadeira de rodas também relataram dificuldades respiratórias significativamente mais frequentes do que todos os outros grupos de mobilidade (0,76±0,43 vs. 0,26±0,44, 0,37±0,49 e 0,31±0,48; p<0,001). As dificuldades respiratórias não diferiram entre os diferentes tipos de RYR1-RD. A análise da diferença entre homens e mulheres resultou em um número total significativamente maior de sintomas para mulheres (10,8 ± 4,1 vs 9,2 ± 4,3; p = 0,004); em particular, as mulheres relataram fraqueza muscular generalizada com mais frequência (0,89 ± 0,32 vs 0,75 ± 0,44, p = 0,006). O modo de herança não teve influência na quantidade de sintomas. Ao correlacionar a fadiga com limitações na vida diária e fraqueza muscular, associações significativas foram encontradas para todos esses sintomas, exceto para dificuldades respiratórias e atrasos nos marcos motores, com dificuldades para correr e levantar tendo a maior correlação com a fadiga. No entanto, essas associações foram de ruins a razoáveis [10] (Tabela 4).
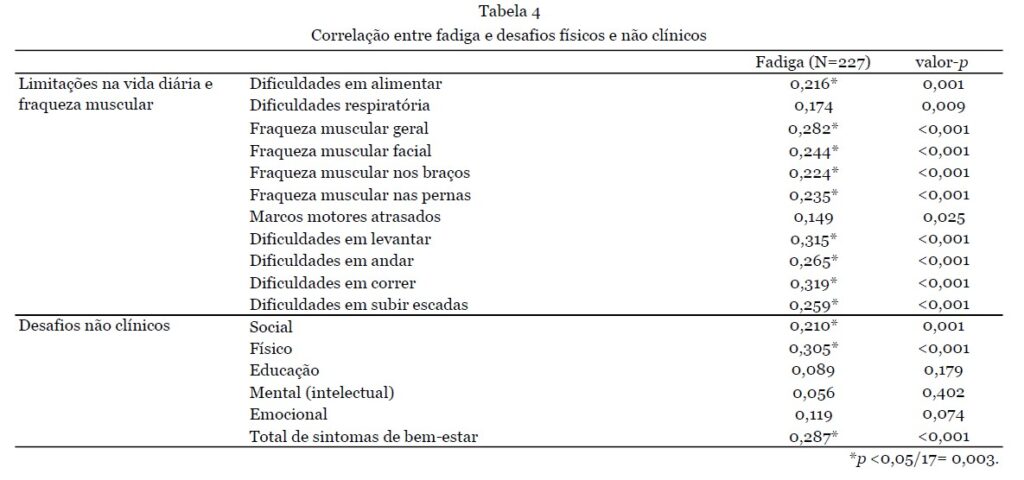
A segunda questão desta parte da pesquisa foi relacionada ao impacto do RYR1-RD no bem-estar geral. A questão "O RYR-1 afeta seu bem-estar em alguma das seguintes áreas" gerou as seguintes respostas: "fisicamente" (n = 202/227, 89%), "emocionalmente" (n = 145/227, 64%), "socialmente" (n = 132/227, 58%), financeiramente (n = 86/227, 38%), nutricionalmente (n = 60/227, 26%), educacionalmente (31/227, 12%) e espiritualmente (n = 18/227, 7%) (Fig. 3C). Além disso, os entrevistados foram solicitados a elaborar esta questão com comentários abertos (n = 168/227); exemplos de cada categoria destes são apresentados na Tabela 5.

Não foram encontradas diferenças entre os diferentes tipos de RYR1-RD (miopatia, MHS ou ambos) para os desafios não clínicos mais frequentemente mencionados (físicos, emocionais e sociais) ou o número total de desafios não clínicos. Ao diferenciar entre os diferentes estados de deambulação, os participantes que necessitam de uso de cadeira de rodas em tempo integral relataram manifestações sociais mais frequentemente e um maior número total de outros desafios não clínicos em comparação com aqueles capazes de andar sem ajuda (0,76 ± 0,43 vs 0,5 ± 0,50; p = 0,017 e 3,9 ± 1,7 vs 2,7 ± 1,6; p < 0,001, respectivamente). As mulheres relataram mais frequentemente seu bem-estar social como adversamente afetado em comparação aos pacientes do sexo masculino (0,64 ± 0,48 vs 0,48 ± 0,50; p = 0,018). Adultos indicaram com mais frequência que seu bem-estar emocional foi afetado negativamente em comparação com crianças (0,71±0,46 vs 0,48±0,50; p=0,001). Ao avaliar a correlação entre esses sintomas e fadiga, algumas associações significativas foram encontradas (Tabela 4) com apenas correlações fracas a razoáveis.
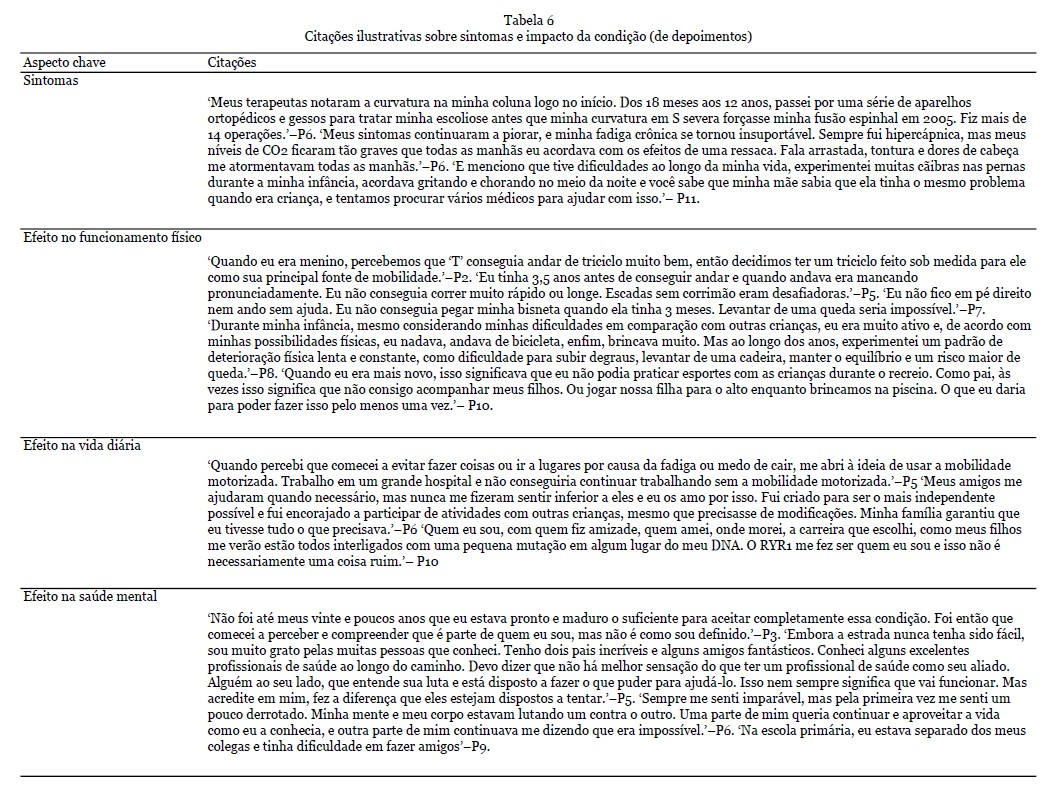
Resultados dos depoimentos - No total, 108 citações foram identificadas neste tema, com uma seleção de citações apresentadas na Tabela 6. Os sintomas mais comuns que surgiram os depoimentos dos pacientes foram físicos (em particular dor e fadiga) e sociais/emocionais, observando as barreiras físicas e mentais que o RYR1-RD introduziu em suas vidas. Muitos indivíduos indicaram um efeito benéfico da atividade regular e do exercício em seus sintomas, ao mesmo tempo em que expressaram preocupações e incertezas sobre quais tipos e frequência de atividades que podem ser mais benéficos, ao mesmo tempo em que tentam evitar dor excessiva e danos musculares. Vários indivíduos perguntaram sobre abordagens nutricionais que podem promover a saúde muscular. Outros compartilharam abordagens que eles experimentaram como benéficas, incluindo suplementação com creatina e magnésio.
Sintomas - Os sintomas de RYR1-RD se apresentaram em alguns pacientes no nascimento e se desenvolveram mais tarde na vida em outros. Os sintomas congênitos incluíam quadris deslocados, pernas fraturadas e dificuldades físicas. Além disso, alguns pacientes tinham má oclusão dentária grave, exigindo cirurgia de mandíbula. Outros pacientes passaram por uma série de aparelhos ortopédicos e operações para tratar escoliose. Além das limitações físicas, sintomas não neuromusculares como fraqueza respiratória e fadiga crônica também foram relatados. Dificuldades de mastigação e/ou deglutição contribuíram para a dificuldade em manter o peso. Ser incapaz de regular a temperatura corporal, sofrer de cãibras nas pernas ou sentir dormência nas pernas foram sintomas adicionais mencionados. Em alguns pacientes, esses sintomas continuaram a piorar ao longo do tempo.
Efeito no funcionamento físico - No geral, os pacientes apresentaram dificuldades físicas significativas. Isso foi expresso principalmente por meio do atraso na obtenção de marcos motores e fraqueza muscular. Por exemplo, ser capaz de andar independentemente era um desafio. À medida que os pacientes envelheciam, os desafios permaneceram para outras atividades físicas, como subir escadas e/ou correr. Outras dificuldades mencionadas incluíam sentar, levantar, curvar-se, amarrar cadarços, ficar de pé por alguns minutos e manter o equilíbrio. A gravidade no funcionamento físico variou entre os participantes. Muitos pacientes relataram não ter conseguido acompanhar seus colegas na infância ou na idade adulta. Alguns (avós) pais expressaram como gostariam de acompanhar seus (netos). A maioria dos participantes usou várias ferramentas para lidar com suas dificuldades físicas. Um dos pais fez um triciclo personalizado para seu filho. Além disso, os pacientes usaram muletas de antebraço, bengalas, cadeiras de rodas e elevadores de cadeira. Embora a grande maioria dos participantes tenha compartilhado suas experiências com incapacidades físicas, alguns participantes mencionaram que haviam alcançado a maioria dos marcos motores e ainda eram capazes de viver uma vida sem dificuldade ou muita adaptação.
Efeito na vida diária - Os participantes enfatizaram o efeito da condição em sua vida diária e como vários sintomas afetaram sua qualidade de vida. Por exemplo, alguns participantes expressaram como a fadiga e o medo de cair impactaram sua vida diária. Enquanto alguns participantes tinham energia suficiente apenas para o trabalho, outros tiveram que se aposentar mais cedo devido à sua condição. A fadiga e a dor também foram relatadas como barreiras à participação social, sendo uma razão para alguns participantes evitarem sair. Um paciente explicou como havia pequenas coisas que eles não conseguiam fazer e que provavelmente seriam consideradas certas por pessoas saudáveis/outros indivíduos. Por exemplo, ser capaz de caminhar certas distâncias, subir um degrau ou lance de escadas e levantar objetos mais pesados. Apesar de suas limitações, os pacientes queriam ser independentes, e as crianças queriam acompanhar seus colegas. A maioria dos pacientes mencionou como eles ainda podem funcionar de forma independente, apesar de sua doença, especialmente com algumas modificações, por exemplo, usando uma cadeira de rodas (motorizada). Simultaneamente, eles também expressaram como eles precisavam de assistência com cuidados e apoio de outras pessoas. Os pacientes explicaram como eles conseguiram ir para a faculdade, se formar, ter uma vida social, cuidar de si mesmos e se sentir realizados e orgulhosos. No entanto, os pacientes também reconheceram como sua condição, pelo menos, os definia em parte, impactando escolhas e como eles sentiam que eram vistos pelos outros. Com a idade, esses desafios se tornaram mais frequentes e impactantes. Para os pacientes, um tratamento eficaz permitiria uma melhora na vida diária e se tornar/permanecer independente.
Efeito na saúde mental - RYR1-RD impactou pacientes e familiares, passando por um longo processo de aceitação da condição. Os pais se sentiam solitários e isolados às vezes, por não conhecerem ninguém em uma posição semelhante. Alguns pacientes falaram sobre as dificuldades que enfrentaram durante a infância. Os pacientes se sentiam envergonhados, por exemplo, sobre suas cicatrizes, e um indivíduo mencionou ter sofrido bullying por causa de um suporte lombar. Em geral, eles achavam difícil fazer amigos. Os familiares sentiam episódios de culpa e também tinham dificuldades em processar a condição. No geral, os pacientes vivenciavam sentimentos de decepção, derrota e tristeza. A sensação de ser diferente causava sentimentos de solidão. Além disso, também foi mencionado como os pacientes se sentiam muito gratos pelo apoio que recebiam da família, amigos e profissionais de saúde. A disposição dos profissionais de saúde em tentar entender e ajudar os pacientes era apreciada. Alguns pacientes mencionaram como sua religião e o apoio da família e dos amigos os fortaleceram. Enquanto um participante explicou a dificuldade de pedir ajuda, outro participante mencionou como amigos e familiares eram um incentivo para participar de atividades e se tornarem independentes. Apesar das dificuldades e impossibilidades vivenciadas pelos pacientes, também foi mencionado como alguns indivíduos tentaram se concentrar em todas as possibilidades que tinham. No entanto, houve uma necessidade de apoio profissional para a saúde mental expressa por pacientes e familiares.
ATIVIDADE FÍSICA
Resultados da pesquisa - Esta parte consistiu em quatro perguntas sobre o número e o tipo de atividade física e a preservação ou melhora da força experimentada. Os participantes relataram a frequência da atividade física como 1-2 vezes por semana (n=66, 29%), 3-4 vezes por semana (n=64, 28%), 5-6 vezes por semana (n=38, 17%) e pelo menos diariamente (n=23, 10%). Apenas 32 pacientes (14%) não participaram de atividade física ou exercício algum. A frequência da atividade física não diferiu entre os diferentes tipos de RYR1-RD. Para os participantes que necessitaram de assistência de cadeira de rodas, uma correlação moderadamente negativa foi encontrada entre a quantidade de exercício e a fadiga (r(12) =–0,622, p=0,023).
Uma ampla gama de atividades físicas específicas foi relatada por 193 pacientes, incluindo caminhada (n = 130/193, 67%), fisioterapia (n = 63/193, 33%), natação (n = 53/193, 27%), treinamento de peso e resistência (n = 39/193, 20%) e ciclismo (n = 33/193, 17%). A maioria dos pacientes relatou ser capaz de manter ou melhorar sua força e/ou resistência (n = 94/197, 48%), enquanto 52 pacientes (26%) não apresentaram melhora. Os outros participantes (n = 51/197, 26%) indicaram que não tinham certeza. Os motivos para não participar de esportes foram "Não consigo criar uma rotina de exercícios que funcione para mim" (23/40, 58%); ‘Estou preocupado com a minha segurança’ (n = 10/40, 25%); ‘Não acredito que o exercício físico irá manter ou melhorar a minha condição’ (n = 9/40, 23%); e ‘Não tenho tempo’ (n = 3/40, 8%).
TRATAMENTO
Resultados dos depoimentos - No total, 20 citações sobre tratamentos anteriores e em andamento foram identificadas. Uma seleção de citações é apresentada na Tabela 7.
Atualmente, não há tratamento definitivo disponível para RYR1-RD. No entanto, várias terapias estavam sendo usadas para ajudar a aliviar os sintomas. Além disso, um quarto dos pacientes relatou como otimizaram sua nutrição e suplementação, por exemplo, tomando creatina em pó para promover massa muscular, bem como várias vitaminas. Um paciente do MHS explicou como eles usaram dantrolene em baixa dosagem e como isso levou a uma qualidade de vida aceitável. Outro paciente usou ventilação não invasiva, permitindo um ganho de energia e um humor geral melhor. A maioria dos pacientes havia iniciado algum tipo de fisioterapia. Em alguns casos, isso foi combinado com terapia ocupacional e terapia da fala. Um paciente mencionou o uso de diferentes tipos de tratamento, como terapia de calor, massagem, ventosaterapia, mas também analgésicos para reduzir dores musculares e espasmos nas costas. Vários pacientes tiveram a oportunidade de participar de um ensaio clínico de medicamentos, com uma experiência geral positiva. Em geral, os participantes descreveram que, embora não se esperasse que o medicamento do estudo curasse a doença, ele tinha sido a única opção para potencialmente melhorar sua QV. Além disso, outro motivador para participar de um ensaio clínico ou tentar vários tipos de suplementos era encontrar um tratamento eficaz para os outros, especialmente para os pais e seus próprios filhos.

PESQUISA E ESTUDOS CLÍNICOS
Resultados da pesquisa - A parte final da pesquisa (cinco perguntas) abordou se os participantes participariam de ensaios clínicos e o que eles achavam que os pesquisadores precisavam saber sobre viver com um RYR1-RD. A questão se os participantes estariam dispostos a participar de um ensaio clínico foi amplamente respondida positivamente ('definitivamente participar' (n = 90/226, 40%), 'provavelmente participará' (n = 102/226, 45%), 'improvável participar' (n = 22/226, 10%) 'não participará' (n = 10/226, 4%). Os participantes que responderam negativamente o fizeram principalmente por preocupações com o impacto na rotina diária (n = 19/32,59%), preocupações com sua segurança (n = 16/32, 50%) e falta de crença em um tratamento ou cura (n = 2/32, 6%).
Com 154 pacientes respondendo a 'o que os pesquisadores precisam saber ou entender melhor sobre viver com RYR-1?', a maioria declarou muitas questões além das preocupações estritamente médicas (incluindo dor) que os pacientes enfrentam frequentemente em sua vida diária. Além disso, os participantes indicaram certas modificações nutricionais e medicamentos que achavam que poderiam ajudar com seus sintomas. Uma seleção dos As respostas a esta questão aberta são apresentadas na Tabela 8. Finalmente, as perguntas dos participantes para a comunidade médica se concentraram principalmente na melhoria do bem-estar físico e emocional (n = 167/210, 80%), no alívio dos sintomas diários (n = 107/210, 51%) e no impacto dos fatores ambientais (n = 129/210, 61%).
EXPECTATIVAS
Resultados dos depoimentos - Identificamos 38 citações sobre expectativas do curso natural da doença e desenvolvimentos em pesquisa. Uma seleção de citações é apresentada na Tabela 9.
Expectativas - Os participantes compartilharam seus pensamentos e expectativas em relação aos tratamentos futuros. Em geral, os participantes esperam um tratamento eficaz, pelo menos retardando a progressão de sua doença. Alguns participantes expressaram o desejo de recuperar a força, a resistência e a possibilidade de poder realizar atividades como correr, subir escadas ou levantar objetos. Outros participantes enfatizaram a motivação de ajudar outros pacientes e famílias, por exemplo, por meio da participação em ensaios clínicos para avançar ainda mais o conhecimento da doença e, ao mesmo tempo, retribuir algo à comunidade científica. Um participante mencionou o desejo de defender os outros e poder entrar em contato com esses pacientes. Vários participantes expressaram a necessidade de uma cura para si próprios e futuros pacientes para tratar sintomas como cãibras musculares, fadiga e oftalmoparesia. A expectativa de declínio da função ao longo do tempo foi percebida como preocupante. Enquanto um participante elaborou sobre a progressão da doença vivenciada, outro participante mencionou o medo de ser pai ou mãe de uma criança afetada e os possíveis desafios para o futuro.
Obtendo conhecimento - Os participantes enfatizaram a importância de obter conhecimento sobre a condição e possíveis tratamentos. A maioria dos participantes conduziu sua própria busca por informações, principalmente por curiosidade, mas também devido à falta de informações suficientes fornecidas por seus provedores de saúde. Além disso, um participante mencionou a importância de informações abrangentes, especialmente para pessoas com formação não científica e devido à grande quantidade de informações disponíveis. A maioria dos participantes expressou o quanto o diagnóstico significou para eles, mesmo com a ausência de cura. Simultaneamente, os participantes também elaboraram sobre o acesso ao suporte. Uma compreensão da doença e saber que a pesquisa está sendo conduzida foi considerada impactante. Poder conhecer pessoas com um diagnóstico semelhante e desafios semelhantes também foi útil. Os participantes tiveram contato com outras pessoas por meio de mídia social, e-mail e videochamadas. Era importante que os pacientes e familiares conhecessem seus pares e pudessem se relacionar com outras pessoas. O papel crucial da organização de pacientes também foi mencionado aqui, principalmente ao fornecer acesso à informação, abordar os desafios e conectar as pessoas. Por fim, muitas pessoas expressaram sua sincera gratidão pelo trabalho dos clínicos e pesquisadores da área, ao mesmo tempo em que os encorajaram a continuar progredindo no desenvolvimento da terapia.

DISCUSSÃO
Por meio da pesquisa realizada neste estudo, coletamos opiniões de pacientes com uma ampla gama de RYR1-RD (n = 227). Os resultados mostram que indivíduos de todas as idades são afetados por RYR1-RD e que a idade no diagnóstico varia amplamente. Pacientes com miopatia foram diagnosticados mais cedo do que aqueles com MHS (mediana de 18,5 e 29,0, respectivamente). A maioria dos pacientes, tanto com miopatia quanto com MHS, era ambulatória e aproximadamente metade dos participantes considerou sua doença progressiva. Com base nos resultados da pesquisa e 12 depoimentos, os pacientes relataram um grande impacto do RYR1-RD em suas vidas. A maioria dos entrevistados relatou estar envolvida em atividade física regular ou exercício, metade dos quais experimentou melhora de força e/ou resistência. Finalmente, a maioria dos pacientes estava disposta a considerar a participação em futuros ensaios clínicos.
Ao refletir sobre essas descobertas, notamos que os tipos relatados de RYR1-RD são semelhantes aos relatados em publicações anteriores na área, com CCD e MHS sendo os mais prevalentes [1,2]. No entanto, este estudo fornece a perspectiva mais abrangente do paciente até o momento sobre o amplo impacto da RYR1-RD na vida cotidiana, fornecendo insights únicos e importantes para consideração futura. Os participantes relataram múltiplos domínios da vida diária influenciados por sua doença: físico, social, emocional e, com menos frequência, financeiro, nutricional e espiritual. Apenas 8% dos participantes relataram que a RYR1-RD não impactou seu bem-estar geral. Essas descobertas estão de acordo com um estudo recente sobre a qualidade de vida relacionada à saúde em indivíduos afetados com RYR1-RD [11]. Os domínios mais valiosos revelados pelas análises qualitativas deste estudo foram a importância dos impactos sociais, o desenvolvimento de estratégias de enfrentamento, tanto físicas quanto psicológicas, e a identificação de fadiga e fraqueza como sintomas principais.
Os achados apoiam observações anteriores de um estudo de questionário em pacientes holandeses com RYR1-RD, demonstrando comprometimentos funcionais substanciais e fadiga crônica em comparação com controles saudáveis [3]. Como esperado, descobrimos que fadiga, dor e dificuldades físicas e sociais associadas foram mais pronunciadas em indivíduos com miopatias congênitas relacionadas ao RYR1 em comparação com indivíduos com MHS. O bem-estar emocional foi mais impactado em adultos do que em crianças. As crianças geralmente foram diagnosticadas em uma idade mais precoce, provavelmente devido ao recente aumento do conhecimento científico e médico sobre RYR1RD. A maioria dos adultos se lembra de saber apenas que algo estava errado quando eram jovens, mas não o que era e como lidar com isso. Para a maioria dos pacientes adultos, seu diagnóstico foi uma longa jornada tortuosa que levou muitos anos e também impactou significativamente outros membros da família.
Além disso, nossos resultados mostraram uma baixa correlação entre fadiga e vários outros sintomas, como fraqueza muscular. No entanto, a fadiga resultou em uma carga significativa em suas vidas diárias. A fadiga é influenciada por vários fatores, direta ou indiretamente [12]. Portanto, as correlações separadas podem ser baixas, mas o impacto na fadiga alto. Concluímos que casos relativamente leves do espectro clínico RYR1-RD estão, no entanto, associados a fadiga grave e limitações funcionais, resultando em perda substancial de qualidade de vida [3]. Por exemplo, a carga diária experimentada por indivíduos com MHS foi confirmada em nosso recente estudo transversal, mostrando uma alta prevalência de sintomas neuromusculares, como mialgia, cãibras musculares e fadiga, resultando em consultas médicas frequentes para identificar a causa desses sintomas [13]. Consistente com isso, a combinação de sintomas neuromusculares e não neuromusculares ocasionalmente piorando ao longo do tempo foi descrita nos depoimentos de alguns pacientes.
Pacientes com RYR1-RD relataram ser fisicamente ativos, frequentemente praticando caminhada, ciclismo, fisioterapia e natação ou hidroginástica, sem diferença na frequência de treinamento entre indivíduos com diferentes tipos de RYR1-RD. Notavelmente, treinamento com pesos ou resistência também foi relatado por 17% de todos os pacientes, atividades geralmente não recomendadas para indivíduos com miopatias [14–16]. Uma ampla gama de outras atividades físicas e esportes foram relatados, incluindo passeios a cavalo, ioga e pilates. Dificuldades em encontrar uma forma adequada de exercício ou preocupações com a segurança foram relatadas como barreiras específicas à participação em atividades físicas. Assim, a orientação sobre formas recomendadas de exercício, fisioterapia e treinamento poderia ser adicionada às diretrizes de cuidados clínicos recentemente desenvolvidas pela RYR-1 Foundation [17]. Descobriu-se que os pacientes faziam uso de várias ferramentas para lidar com suas lutas físicas. A função física diferiu entre os pacientes, e alguns expressaram como sentiam que ainda poderiam viver uma vida sem dificuldade ou muita adaptação. No entanto, a perda de força e resistência ao longo do tempo pode ser uma barreira significativa para vários tipos de atividades. Os pacientes estavam cientes do impacto emocional de sua doença e, portanto, precisam de recursos para ajudá-los a lidar com esses sentimentos, algo especialmente vivenciado pelos pais. Uma mistura semelhante de respostas emocionais também foi relatada nos depoimentos. Embora alguns pacientes tenham mencionado aspectos emocionais desafiadores associados à vida com um RYR1RD, o apoio da família, amigos e profissionais de saúde os ajudou a lidar com esses sentimentos.
Finalmente, este estudo também ofereceu uma perspectiva muito útil e única do paciente sobre futuros ensaios clínicos. A maioria dos participantes indicou que eles estavam (provavelmente) dispostos a participar, com as principais barreiras à participação sendo o impacto antecipado nas atividades da vida diária e preocupações com a segurança. Esta informação é importante, pois a perspectiva do paciente e a consideração de potenciais barreiras à participação são altamente relevantes para o desenho de ensaios clínicos e estão sendo cada vez mais consideradas. Embora as perspectivas já tenham sido fornecidas por grupos de defesa do paciente para atrofia muscular espinhal [5, 18], esta contribuição valiosa ainda está faltando para muitas outras condições neuromusculares. Alguns participantes também relataram suas experiências relacionadas a ensaios clínicos em andamento ou concluídos. Pesquisas futuras devem se concentrar na perspectiva do paciente em relação às expectativas terapêuticas e ao desenho de ensaios clínicos, o que pode melhorar significativamente o recrutamento, a retenção e os resultados do estudo [5].
Este estudo tem várias limitações. Primeiro, como a participação na pesquisa foi anônima, o diagnóstico foi relatado pelos pacientes e não pôde ser verificado de forma independente pelos clínicos que os diagnosticaram. Esta foi uma escolha deliberada dos pacientes que iniciaram esta pesquisa para reduzir o viés de averiguação, não limitando a participação a indivíduos com contato recente com profissionais de assistência médica. Segundo, o desenho do estudo inclui um viés de averiguação para indivíduos com acesso à internet e que foram informados sobre a RYR-1 Foundation e suas atividades. Terceiro, a pesquisa consistiu apenas em perguntas personalizadas. Os pesquisadores consideraram adicionar questionários validados sobre habilidades funcionais ou fadiga, mas deliberadamente escolheram não fazê-lo e se concentrar nas questões levantadas pela comunidade de pacientes. Sendo um questionário não validado, as formulações de certas perguntas podem ter levado a diferentes interpretações, possivelmente impactando os resultados. Por exemplo, esperava-se que as dificuldades respiratórias fossem mais frequentes para pacientes com miopatia congênita em comparação com MHS. No entanto, isso não foi encontrado nos resultados da pesquisa, o que pode refletir diferentes interpretações dos participantes sobre o que constitui dificuldades respiratórias. Se uma pergunta sobre o bem-estar dos participantes não revelar um problema específico, isso não significa necessariamente que o aspecto específico do bem-estar não seja diferente em comparação com indivíduos sem miopatia subjacente ou RYR1-RD. Por exemplo, se os participantes fizessem alterações em seu programa escolar, seu bem-estar educacional poderia ser como o de indivíduos sem miopatia subjacente ou RYR1-RD porque eles podem ir à escola como acharem adequado. No entanto, as perguntas usadas nesta pesquisa fornecem uma ideia geral de como os participantes são impactados por sua doença. Finalmente, a pesquisa estava disponível apenas em inglês, o que pode ter resultado em uma população de estudo que não é globalmente representativa.
Um ponto forte importante deste estudo é que a pesquisa foi realizada como um esforço colaborativo entre a comunidade de pacientes, uma organização de defesa de pacientes (RYR-1 Foundation) e clínicos-pesquisadores. As questões abordadas são tópicos importantes para a comunidade de pacientes. O design e a execução da pesquisa, bem como a apresentação dos resultados da pesquisa e depoimentos dos pacientes durante o workshop, foram apoiados e facilitados pela RYR-1 Foundation e os resultados foram avaliados quantitativa e qualitativamente em consulta com pesquisadores acadêmicos. No entanto, este estudo foi conduzido principalmente por pacientes e famílias do RYR1-RD, o que resultou em uma participação robusta, um aspecto cuja importância é cada vez mais reconhecida pelas autoridades regulatórias e de reembolso.
Concluindo, este estudo fornece uma perspectiva única do paciente sobre a jornada diagnóstica, o impacto da doença, os desafios psicossociais, as atividades físicas, bem como as expectativas do paciente para tratamentos futuros e ensaios clínicos de indivíduos com RYR1-RD.
AGRADECIMENTOS
Os autores gostariam de agradecer à equipe do RYR-1 Foundation Study que projetou e executou o estudo e a todos os participantes deste estudo pelo tempo dedicado a concluir a pesquisa e por compartilhar seus depoimentos. Vários autores desta publicação são membros do Netherlands Neuromuscular Center (NL-NMD) e da European Reference Network for rare neuromuscular diseases (EURO-NMD).
FINANCIAMENTO
Este trabalho foi apoiado financeiramente pelo Princes Beatrix Fund (número de concessão W.OR22-10) e pelos Institutos Nacionais de Saúde (número de concessão AR078000).
CONFLITO DE INTERESSE
Heinz Jungbluth é um membro do Conselho Editorial deste periódico, mas não esteve envolvido no processo de revisão por pares nem teve acesso a nenhuma informação sobre sua revisão por pares. Os outros autores não relatam mais conflitos de interesse.
DECLARAÇÃO DE DISPONIBILIDADE DE DADOS
Devido à natureza desta pesquisa, os participantes deste estudo não concordaram que seus dados fossem compartilhados publicamente, portanto, dados de apoio não estão disponíveis.
MATERIAIS SUPLEMENTARES
O material suplementar está disponível na versão eletrônica deste artigo: https://dx.doi.org/ 10.3233/JND-240029.
REFERÊNCIAS
[1] Snoeck M,vanEngelen BG, K¨ usters B, Lammens M, Meijer R, Molenaar JP, et al. RYR1-related myopathies: A wide spectrum of phenotypes throughout life. Eur J Neurol. 2015;22(7):1094-112.
[2] Lawal TA, Todd JJ, Meilleur KG. Ryanodine receptor 1related myopathies: Diagnostic and therapeutic approaches. Neurotherapeutics. 2018;15(4):885-99.
[3] van Ruitenbeek E, Custers JAE, Verhaak C, Snoeck M, Erasmus CE, Kamsteeg EJ, et al. Functional impairments, fatigue and quality of life in RYR1-related myopathies: A questionnaire study. Neuromuscul Disord. 2019;29(1): 30-8.
[4] Ramos-Platt L, Elman L, Shieh PB. Experience and perspectives in the US on the evolving treatment landscape in spinal muscular atrophy. Int J Gen Med. 2022;15: 7341-53.
[5] GussetN,StalensC,StumpeE,KlouviL,MejatA,Ouillade MC, de Lemus M. Understanding European patient expectations towards current therapeutic development in spinal muscular atrophy. Neuromuscul Disord. 2021;31(5):41930.
[6] O’ConnorTN,vandenBersselaarLR,ChenYS,NicolauS, Simon B, Huseth A, et al. RYR-1-related diseases international research workshop: From mechanisms to treatments pittsburgh, PA, U.S.A., 21-22 July 2022. J Neuromuscul Dis. 2023;10(1):135-54.
[7] Priest H, Roberts P, Woods L. An overview of three different approaches to the interpretation of qualitative data. Part 1: Theoretical issues. Nurse Res. 2002;10(1): 30-42.
[8] Vaismoradi M, Turunen H, Bondas T. Content analysis and thematic analysis: Implications for conducting a qualitative descriptive study. Nurs Health Sci. 2013;15(3): 398-405.
[9] Braun V, Clarke V. Using thematic analysis in psychology. Qualitative Research in Psychology. 2006;3(2):77-101.
[10] Akoglu H. User’s guide to correlation coefficients. Turk J Emerg Med. 2018;18(3):91-3.
[11] Capella-Peris C, Cosgrove MM, Chrismer IC, EmileBacker M, Razaqyar MS, Elliott JS, et al. Mixed methods analysis of Health-Related Quality of Life in ambulant individuals affected with RYR1-related myopathies pre-post-N-acetylcysteine therapy. Qual Life Res. 2020;29(6):1641-53.
[12] Kalkman JS, Schillings ML, Zwarts MJ, van Engelen BG, Bleijenberg G. The development of a model of fatigue in neuromuscular disorders: A longitudinal study. J Psychosom Res. 2007;62(5):571-9.
[13] van den Bersselaar LR, Jungbluth H, Kruijt N, Kamsteeg E-J, Fernandez-Garcia MA, Treves S, et al. Neuromuscular symptoms in patients with RYR1-related malignant hyperthermia and rhabdomyolysis. Brain Communications. 2022;4(6).
[14] VoetNBM.Exerciseinneuromusculardisorders:Apromising intervention. Acta Myol. 2019;38(4):207-14.
[15] Voet NB, van der Kooi EL, van Engelen BG, Geurts AC. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst Rev. 2019;12(12): Cd003907.
[16] AfridiA,RathoreFA.Whataretheeffects of strength training and aerobic exercise training for muscle disease?- A Cochrane Review summary with commentary. J Rehabil Med. 2021;53(9):jrm00231.
[17] RYR1 Foundation. Chapter 9: Physical Activity and Physical Therapy (PT); 2022 [cited 2022 Nov]. Available from: https://ryr1.org/online-resources-category/physicalactivity-physical-therapy.
[18] McGrawS,Qian Y, Henne J, Jarecki J, Hobby K, Yeh WS. Aqualitative study of perceptions of meaningful change in spinal muscular atrophy. BMC Neurol. 2017;17(1):68.
Buscando estar sempre conectado com as notícias relacionadas e de interesse dos portadores de mutação no gene RYR1, mais especificamente a Miopatia Congênita Centronuclear, que se trata da doença que me acomete, tomei conhecimento da recente pré-publicação científica, intitulada, “O Propofol liga-se diretamente e inibe o receptor 1 de rianodina do músculo esquelético (RYR1)”, versão postada em 12 de janeiro de 2024, na qual os pesquisadores, Thomas T. Joseph, MD, PhD¹, Weiming Bu, PhD¹, Omid Haji-Ghassemi, PhD², Yu Seby Chen, PhD², Kellie Woll, PhD², Paul D. Allen, MD, PhD⁴, Grace Brannigan, PhD³, Filip Van Petegem, PhD², Roderic G. Eckenhoff, MD¹, através de resultados obtidos em estudos e ensaios sugerem em seus relatos que o propofol, um agente anestésico intravenoso de curta ação, em concentrações clínicas, se liga ao receptor de rianodina tipo 1 (RYR1), inibindo sua abertura, podendo assim, prevenir as manifestações clínicas da Hipertermia Maligna (HM), mesmo com exposição a agentes desencadeantes como os anestésicos voláteis.
Confira a integra da publicação pelo seguinte link: https://www.biorxiv.org/content/10.1101/2024.01.10.575040v1.full
O receptor de rianodina tipo 1 (RYR1) desempenha um papel central na determinação de quando (tempo), e quanta (quantidade) força é produzida pelos músculos esqueléticos, que é necessária e essencial para movimentação do corpo e atividades físicas diárias dos indivíduos. Como principal canal de liberação de íon de cálcio (Ca²⁺) no retículo sarcoplasmático do músculo esquelético, a mutação genética no receptor de rianodina tipo 1 (RYR1), tem a ela subjacentes, algumas doenças ou distúrbios musculares, tais como a Miopatia Centronuclear, Miopatia Central Core, Miopatia Multi-Minicore, Desproporção Congênita do Tipo de Fibra, incluindo distúrbios como a Rabdomiólise por Esforço, e a Hipertermia Maligna (HM).
Em pacientes com a mutação no gene RYR1, “a crise” de Hipertermia Maligna, causada pela exposição a algumas drogas desencadeantes, como os anestésicos voláteis halogenados, pode direcionar o RYR1 a deixar o canal de rianodina em um estado aberto, resultando em uma liberação descontrolada de Ca²⁺, acarretando em tensão no sarcômero, e consequente produção de calor. A restauração de Ca²⁺ no retículo sarcoplasmático também consome ATP (adenosina trifosfato), molécula responsável pelo depósito de energia celular, gerando também por consequência uma carga metabólica adicional insustentável.
Ao anestesiar pacientes com mutações genéticas conhecidas pela suscetibilidade a Hipertermia Maligna, o anestésico geral intravenoso não desencadeante propofol é comumente substituído por anestésicos desencadeantes. As evidências de ligação direta de agentes anestésicos ao RYR1 ou seus parceiros de ligação são escassas, e as interações em nível atômico do propofol com o RYR1 são totalmente desconhecidas. Os pesquisadores mostram no trabalho acima descrito que o propofol diminui a abertura do receptor do canal de rianodina (RYR1) com vesículas no retículo sarcoplasmático e bicamadas lipídicas planas, e que inibe a liberação de Ca²⁺ induzida por ativador do retículo sarcoplasmático no músculo esquelético humano. Além de confirmar a ligação direta, a marcação por fotoafinidade usando m-azipropofol (AziPm) revelou vários supostos locais de ligação de propofol no RYR1. A projeção pela simulação dinâmica da afinidade de ligação molecular sugere que o propofol se liga a pelo menos um destes locais em concentrações clínicas. Esses achados convidam à hipótese de que, além de o propofol não desencadear a Hipertermia Maligna, ele também pode ser protetor contra a Hipertermia Maligna, inibindo o fluxo induzido de Ca²⁺ através do canal de rianodina - RYR1.
¹ Department of Anesthesiology and Critical Care, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA USA; ² Department of Biochemistry, University of British Columbia, Vancouver, BC, Canada; ³ Department of Physics and Center for Computational and Integrative Biology, Rutgers University, Camden, NJ USA; ⁴ Department of Anesthesiology, University of Tennesee, Knoxville, TN USA
O artigo acima mencionado foi publicado na bioRxiv, repositório aberto de pré-publicação direcionado as ciências biológicas (https://www.biorxiv.org/), e hospedado pelo Cold Spring Harbor Laboratory (CSHL).
As doenças relacionadas ao RYR1 são identificadas com base em sua classificação histopatológica, isto é, pela aparência da biópsia do músculo na lâmina do microscópio. A diferenciação encontrada na biópsia designará o tipo da doença, se é por exemplo, Miopatia Central Core, Miopatia Multiminicore, Miopatia Centronuclear, ou Desproporção Congênita de Tipos de Fibras.
A Miopatia Congênita Centronuclear (MCCN) e a Miopatia Central Core (MCC) são ambas doenças musculares hereditárias causadas por mutações genéticas. A MCCN pode estar associada a mutações em diferentes genes, no DNM2, BIN1, MTM1, e RYR1, já a MCC está associada somente ao gene RYR1.
Esses tipos de doenças relacionadas ao RYR1 variam amplamente em termos dos seus diferentes sinais e sintomas, de quando eles inicialmente se apresentaram, além da sua respectiva gravidade. Embora sejam altamente variáveis, os sintomas presentes também dependem se a mutação do gene RYR1 é autossômica dominante ou autossômica recessiva.
Uma pergunta que sempre chega até mim é sobre as diferenças entre a Miopatia Centronuclear e a Miopatia Central Core. Assim, eu, enquanto portador da Miopatia Congênita Centronuclear (MCN), buscarei esclarecer pontualmente neste texto, me atendo ao tipo que me acomete, que é a pela mutação no gene RYR1.
Estas doenças apesar de terem sintomas parecidos e compartilharem de algumas características clínicas em comum, se confundem entre si, e apresentam com algumas diferenças distintas:
Miopatia Congênita Centronuclear (MCCN)
Miopatia Central Core (MCC)
Em resumo, a Miopatia Congênita Centronuclear e a Miopatia Central Core, ambas doenças relacionada ao RYR1, têm seus sintomas e sinais físicos que podem se parecer, podem se confundir, mas são diferentes, a contar da análise histológica das células musculares em uma biópsia, exame este que é crucial para diferenciar entre as duas condições e determinar o diagnóstico correto, conduta médica, tratamento, e até prognóstico de evolução.
Nota: todo o material escrito nesta página é oriundo de pesquisa científica, e conclusões próprias do autor deste website, que convive como portador, desde o nascimento, com a Miopatia Congênita Centronuclear causada pela mutação no gene RYR-1
ARMGO PHARMA PÚBLICA IMPORTANTES E POSITIVOS RESULTADOS DO ENSAIO DE FASE 1B DO RYCALl® ARM210 PARA O TRATAMENTO DE MIOPATIAS RELACIONADAS AO RECEPTOR DE RIANODINA 1
ARMGO Pharma, Inc. (ARMGO), uma empresa privada no setor biofarmacêutico que desenvolve uma nova classe de drogas de moléculas pequenas ou micro moléculas conhecidas como Rycals®, anunciou em 29 de janeiro de 2024 a publicação dos resultados de um estudo de Fase 1b de seu Rycal ARM210 (também conhecido como S48168), para o tratamento de miopatias relacionadas ao receptor 1 de Ryanodina (RYR1-RM), uma doença muscular órfã, também chamada de “doença rara”, por ser uma doença que afeta uma pequena percentagem da população.
2024 a publicação dos resultados de um estudo de Fase 1b de seu Rycal ARM210 (também conhecido como S48168), para o tratamento de miopatias relacionadas ao receptor 1 de Ryanodina (RYR1-RM), uma doença muscular órfã, também chamada de “doença rara”, por ser uma doença que afeta uma pequena percentagem da população.
Os dados foram publicados em um artigo intitulado 'Rycal S48168 (ARM210) para Miopatias Relacionadas ao RYR1: um ensaio de fase um, estudo em aberto, e de ensaio de escalonamento de dose', de autoria do Dr. Joshua Todd et al, no Journal eClinicalMedicine, parte da família de publicações Lancet. O artigo revisa os dados do estudo de Fase 1b do ARM210 e seu novo mecanismo de ação alostérico (MoA) visando a causa raiz da doença relacionada ao RYR-1 (RYR1-RM): mutação do Receptor 1 de Ryanodina (RYR1).
O gene RYR1 que codifica o Receptor 1 de Ryanodina RYR1, um canal intracelular de liberação de cálcio, vaza em doenças musculares. Vazamentos intracelulares de cálcio causados por canais RYR1 com mutação prejudicam a contração muscular, levando à fraqueza muscular e perda de função, e ativam vias tóxicas que danificam os músculos, causando os sintomas das doenças relacionadas ao RYR1.
O ensaio de Fase 1b, aberto, de escalonamento de dose confirmou a segurança, tolerabilidade e farmacocinética da dosagem de 120 e 200 mg de ARM210 diariamente durante 29 dias em homens e mulheres adultos afetados pelas doenças relacionadas ao RYR1 (RYR1-RM).
É importante ressaltar que o estudo também demonstrou eficácia preliminar no grupo de dose mais alta em dois sintomas característicos das doenças relacionadas ao RYR1 (RYR1-RM): 1) alívio significativo da fadiga avaliada pelo sistema PROMIS-fatigue (Patient-Reported-Outcome Measurement Information System) t-scores, e 2) melhora da força dos proximais avaliada pelo exame físico de abdução do ombro (Medical Research Council Grading). Estes resultados justificam o desenvolvimento futuro do ARM210 como um potencial tratamento e modificador da doença para as miopatias relacionadas ao RYR1 (RYR1-RM) em um ensaio de Fase 2 randomizado e controlado por placebo.
O ensaio de Fase 1b concluído foi conduzido em colaboração com o National Institute of Neurological Disorders and Stroke (NINDS) e National Institute of Health (NIH) sob um Acordo Cooperativo de Pesquisa e Desenvolvimento (CRADA - Cooperative Research and Development Agreement ), com o apoio da Fundação RYR-1, Pittsburgh, PA, EUA.

(Foto esquerda para direita: Dr. Tokunbor Lawal (autor da publicação), Dr. Mike Goldberg (co-fundador/Co-presidente de Pesquisas da Fundação RYR-1), e Dr. Payam Mohassel (Pesquisador Principal e Autor Senior)).
“Estamos muito satisfeitos com os resultados do ensaio com as doenças relacionadas ao RYR1 (RYR1-RM) conduzido em conjunto com o NIH, pois o estudo confirmou a segurança e tolerabilidade do ARM210, mas o mais importante, demonstrou pela primeira vez que o nosso Rycal®, ARM210, pode reverter os sintomas desta doença muscular crônica e devastadora em um curto período de tratamento. Isso é muito promissor”, disse Gene Marcantonio, M.D., Ph.D., CEO da ARMGO Pharma. “Esperamos, portanto, continuar rapidamente no desenvolvimento do ARM210 para levar este primeiro e potencial tratamento aos pacientes com as doenças relacionadas ao RYR1, com o apoio da Fundação RYR-1 e da comunidade de pacientes.”
Michael F. Goldberg, MD, MPH, co-presidente de pesquisa da Fundação RYR-1 acrescentou: “Estamos entusiasmados com a publicação deste importante estudo, pois representa um farol de esperança para muitos indivíduos e famílias de todo o mundo afetados pelas doenças relacionadas ao RYR1. Estamos ansiosos pelas próximas etapas de ensaios no desenvolvimento dessa importante droga.”
Mais informações sobre este estudo de Fase 1b podem ser encontradas online em: https://clinicaltrials.gov/study/NCT04141670. O ensaio foi apoiado pelos Programas de Pesquisa Intramural do NIH/NINDS, NIH/NINR, um NIH Clinical Center Bench to Bedside Award (2017-551673) e pelo parceiro de colaboração anterior da ARMGO, Les Laboratoires Servier. O conteúdo é de responsabilidade exclusiva dos autores e não representa necessariamente a opinião oficial dos Institutos Nacionais de Saúde.
BREVE REVISÃO SOBRE A FUNÇÃO DO RYR1 E DOENÇAS RELACIONADAS
Os RYRs são canais homotetraméricos de liberação de cálcio intracelular responsáveis pelo fluxo de Ca2+ dos retículos sarcoplasmáticos/endoplasmáticos (RS/RE) para o citoplasma da maioria dos tipos de células. RYR1 é a isoforma predominante no músculo esquelético de mamíferos, onde a liberação de Ca2+ via RYR1 é necessária para o acoplamento excitação-contração e função muscular normal. Já o RYR2 é a isoforma predominante no músculo cardíaco onde a liberação de Ca2+ via RYR2 é necessária para a função normal do músculo cardíaco. Mutações genéticas humanas nos genes RYR1 e RYR2 fazem com que o cálcio vaze dos canais RYR, levando à doença (Figura 1).
Os canais RYR normalmente alternam entre um estado de repouso (fechado) e excitado (aberto). Em certas doenças, em que existe a mutação genética, o RYR é modificado e torna-se incontinente ou seja, fica vazando. No caso do RYR1, este desempenha um papel crítico no músculo esquelético, e as mutações do RYR1 em humanos levam a uma miopatia progressiva, conhecida como doenças relacionadas ao RYR1 (RYR1-RM), privando o músculo da capacidade de responder eficazmente aos sinais de contração, levando à fraqueza muscular.
Sobre o Rycals® - A inibição do canal interromperia o vazamento, mas esta intervenção geralmente não seria benéfica, uma vez que bloquearia a função normal do RYR. O Rycals®, são moléculas que podem restaurar a função normal do canal sem bloquear o RYR, e abrem a possibilidade de intervenções terapêuticas.
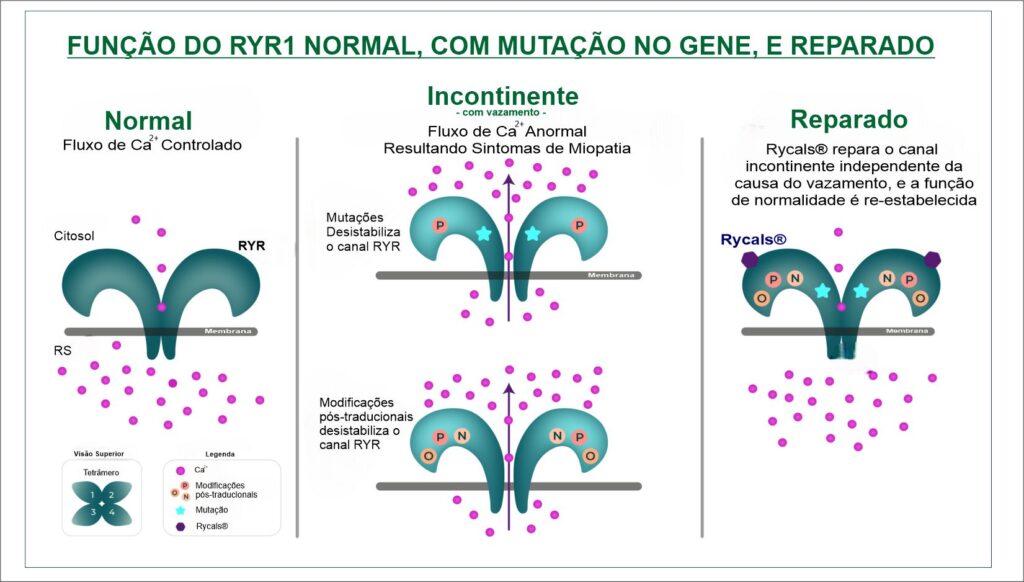
Figura 1: Modelo da função do RYR. O RYR controla o fluxo de cálcio (Ca2+) de dentro do retículo sarcoplasmático (RS) através da membrana até o citoplasma. (Quadro Esquerdo) O RYR normal regula o fluxo de cálcio alternando entre um estado fechado e aberto. (Quadro Meio) Na RYR1-RM, o RYR mutado apresenta vazamento, levando ao fluxo anormal de cálcio, resultando em sintomas da doença. Modificações pós-traducionais* (MPTs) do RYR agravam ainda mais o vazamento. Em outros distúrbios musculares, como DMD, insuficiência cardíaca ou sarcopenia, as modificações pós-traducionais (MPTs) do RYR podem causar vazamento no canal, levando a um fluxo anormal de cálcio, resultando em sintomas da doença. (Quadro Direito) Rycals® liga-se aos canais incontinentes (com vazamento) e repara o vazamento independentemente da causa do vazamento, restaurando a função normal do canal. O Rycals® não bloqueia o RYR.
*As modificações pós-traducionais (MPTs) são modificações químicas e estruturais de uma cadeia proteica após a sua tradução. Estas modificações podem determinar a atividade, a localização e interações com outras proteínas.
Me chamo Lise, e sou mãe de um menino de 10 anos, João Miguel, diagnosticado com a Mutação do Gene Ryrr.1 Central Core. Moramos na cidade de Criciúma no Estado de Santa Catarina, Brasil. Eu e meu esposo esperamos o João por sete anos, e seu nascimento trouxe alegria a nossa casa.

O João nasceu com um pezinho torto congênito, e nos seus primeiros dias de vida passou a usar gesso até os 11 meses de idade, como também foram feitas duas cirurgias para concluir a correção do pé. O João começou a engatinhar com um ano e três meses e caminhar com um ano e nove meses, achávamos que esse atraso motor era pelo tempo que ele passou em recuperação do pezinho.
Aos três anos percebemos as quedas constantes e levamos esta nossa preocupação ao ortopedista que o acompanhava desde bebê. Após analise clinica o Dr percebeu a fraqueza muscular e que o João realizava a manobra de Gowers ao levantar-se. Saímos do consultório com encaminhamento para o Neurologista e ali iniciava-se anos de buscas e angustias por um diagnóstico. Foi realizado a eletromiografia onde constatou-se a fraqueza muscular, e vários exames. Mesmo o CPK com resultado normal, os médicos procuravam as doenças degenerativas mais graves, como Duchenne e Becker.
Aos cinco anos foi feito o exame do Painel Genético para as setenta doenças degenerativas musculares e veio o resultado negativo para todas elas. Foi um misto de alegria e angustia, pois não tínhamos fechado o diagnóstico. Aos três anos iniciamos os tratamentos recomendado pelo médico, com fisioterapias, equoterapia e natação. O desenvolvimento após inicio do tratamento foram visíveis, o João passou a ter mais equilíbrio, as quedas diminuíram, começou a subir escadas com apoio e ter mais firmeza para desce-las, a cada dia percebemos o progresso (lento), mais sempre constante na sua condição física.
Este ano (2023), após 7 anos de busca, conseguimos fechar o diagnóstico do João com o Exoma. Com o diagnostico a fisioterapeuta que o acompanha desde o inicio da nossa jornada montou um novo cronograma de exercícios, e adotamos que a “Vida é Movimento” então vamos nos movimentar e continuar escrevendo uma história linda para nosso menino que é cheio de vida e alegria!
O exame clínico é a técnica médica mais antiga utilizada na obtenção de informações gerais. Ele leva à compreensão sobre a situação do paciente atendido e assim pode-se diagnosticar e direcionar o melhor tratamento.
Na neurologia, atualmente, por mais que tenhamos disponíveis modernos meios de diagnóstico, o exame clínico, incluindo histórico de saúde e sinais físicos, ainda se faz como uma importante ferramenta aos médicos no diagnóstico de distúrbios neurológicos.
Trazendo esse tema para minha realidade, lembro-me de um fato que vivenciei em meados dos anos 60 que marcou muito a minha memória. Acredito ter sido de fundamental importância para o início do processo de investigação diagnóstica da minha patologia, por mais que a palavra conclusiva, de que era portador de Miopatia Congênita Centronuclear, só tenha sido dita 40 anos mais tarde.
Naquela época, com 5 anos de idade, meus pais já haviam peregrinando por vários médicos em busca de diagnóstico. Viviam com questionamentos e dúvidas quanto a minha situação física apresentada desde meu nascimento. Resolveram, então, me levar à consulta com um importante neurologista do Brasil.
Os recursos para diagnóstico disponíveis na época, assim como o conhecimento médico acerca de algumas doenças, eram bem limitados. Contudo, o exame clínico era um procedimento de que os médicos podiam se valer.
Quando disse que aquela consulta ficou marcada na minha memória, lembro que foi tudo meio traumático, porque, na ocasião, o médico pediu que eu me deitasse no chão e, em seguida, que me levantasse e que me colocasse de pé … pedido muito estranho, autoritário, e tarefa fisicamente difícil para mim. Senti que meus pais ficaram também desentendidos com aquela situação.
No final da consulta, o médico deu seu parecer aos meus pais, que acredito ter sido as primeiras palavras sobre o que vinha ser um diagnóstico, ou seja, de que eu era portador de uma “doença muscular”. Isso aconteceu graças àquele teste de me levantar. Tratava-se do sinal clínico chamado de SINAIS DE GOWERS, também conhecido como manobra do levantar miopático.
A fraqueza da musculatura proximal em uma criança, muitas vezes é associada a uma doença neuromuscular e pode ser avaliada pela MANOBRA DE GOWERS.
Os músculos proximais são os músculos que se encontram próximos do tronco, incluindo a parte superior das pernas, quadril, a parte superior dos braços, os ombros e o próprio tronco.
Na Manobra de GOWERS, pede-se que a criança se deite no chão com a barriga para cima e levante até se colocar de pé. A manobra é positiva (sinalizando fraqueza muscular proximal) quando a criança se apoia no chão, e literalmente precisa escalar o próprio corpo, ou as coxas para se levantar (ver figura).

Manobra de Gowers - 1). A criança deitada com a barriga para cima, para se levantar, dado a sua fraqueza muscular, 2). precisa rolar, se colocando de bruços, estendendo seus braços e pernas separados, 3). com a maior parte do peso do tronco apoiando nos braços estendidos, se empurra o corpo para trás para transferir o peso do tronco para as pernas estendidas. 4). Para estender o quadril, a criança coloca as mãos nos joelhos, 5). e sobre os braços pelas coxas, 6). como se estivesse escalando o próprio corpo, 7). até ficar de pé.
A Manobra de Gowers são sinais de movimentação física descritos e deixado como contribuição para a ciência neurológica, oriundo de observações feitas por Sir William Gowers. Gowers, foi um neurologista, Inglês do final do século XIX, que passou sua vida profissional trabalhando no National Hospital for the Relief and Cure for the Paralyzed and Epileptic e no University College Hospital na Queen Square, em Londres, Reino Unido, e após suas próprias observações clínicas, deixou registrado em seu livro Manual of the Diseases of the Nervous System, a descrição dos Sinais de Gowers, composto por uma sequência de sinais observados durante movimentação física, e que eventualmente poderá indicar fraqueza muscular proveniente de uma patologia neuromuscular.
Por fim, gostaria de terminar esse texto destacando os Sinais de Gowers por sua grande importância no exame clínico durante a consulta médica e eventual diagnóstico de uma doença muscular.
Dado à seriedade deste assunto, devo iniciar o texto com as palavras conclusivas sobre o tema… a HIPERTERMIA MALIGNA é uma doença grave, com risco de morte, que se apresenta em forma de crise, acionada por um gatilho, que é normalmente uma droga anestésica, sendo os portadores de mutação no gene RYR1, os indivíduos com maior risco de ser afetado, daí a afirmação, “HIPERTERMIA MALIGNA, PONTO DE ATENÇÃO PARA OS MÉDICOS ANESTESISTAS, PREOCUPAÇÃO PARA OS PORTADORES DE MUTAÇÃO DO GENE RYR1”. Os anestesistas devem ter o conhecimento sobre a doença e assumir que todas as pessoas com mutação no gene RYR1 correm risco de Hipertermia Maligna. No caso de qualquer procedimento médico que seja necessário anestesia, o portador de mutação de no gene RYR1 deve fazer com que o cirurgião e anestesista saiba sobre sua mutação no gene RYR1, portanto com Suscetibilidade a Hipertermia Maligna (MHS), assumindo assim os eventuais riscos, para tomar precauções e administrar um tipo seguro de anestésico. É recomendado também aos portadores da mutação do RYR1 que portem uma identificação de advertência médica indicando seu risco de HM, em caso de emergência, neste caso, um adesivo em todos os documentos pessoais (Ex.: Identidade, CNH, Passaporte, Carteira do Plano de Saúde, e etc).
CONCEITO
A Hipertermia Maligna (HM) é uma patologia de relação farmacogenética. E isso significa que a doença se manifesta através de um episódio/crise em indivíduos que tenham uma suscetibilidade genética, devido a uma mutação em um determinado gene, acontecendo caso sejam expostos a gatilhos anestésicos (fármacos = medicamentos). Essa predisposição é chamada de "Suscetibilidade à Hipertermia Maligna” (MHS). Genericamente, explica-se que a Hipertermia Maligna (HM) é uma reação biológica em que o corpo humano superaquece a ponto de um colapso muscular, e é considerada uma emergência médica. Se alguém com Hipertermia Maligna não for tratado a tempo, como consequência pode resultar em insuficiência renal, dano cerebral, parada cardíaca, falência de órgãos adicionais, e até morte.
Os indivíduos com Suscetibilidade à Hipertermia Maligna (MHS) podem também apresentar uma crise em resposta a outros gatilhos externos, como por exemplo, ao esforço físico, que poderá causar a Rabdomiólise (quebra muscular), e neste caso apresentando outros sintomas, tais como, cãibras severas, rigidez muscular, e intolerância ao calor.
A genética à Suscetibilidade à Hipertermia Maligna (MHS) é complexa, e vários genes têm sido identificados desempenhando um papel patogênico na Hipertermia Maligna, sendo o RYR1 o mais estudado. Este gene codifica o receptor 1 de Ryanodina (RyR-1), uma proteína do canal de cálcio do retículo sarcoplasmático, expressa predominante na célula muscular. Sabe-se que na maioria dos casos da Suscetibilidade à Hipertermia Maligna (MSH), ela apresenta um traço autossômico dominante, e isso significa que se você tem MHS, um de seus pais provavelmente também tem a MHS. Também significa que cada um de seus filhos têm 50% de chance de herdar a MHS. No entanto, a dita complexidade se prova quando os médicos também observaram a Hipertermia Maligna em pessoas com mutações RYR1 autossômicas recessivas.
Em alguns casos, ao contrário do que se pensa, a Suscetibilidade à Hipertermia Maligna (MHS) pode ocorrer na ausência de fraqueza muscular, em outras palavras, os indivíduos com MHS têm força normal ou até mesmo aumentada, e seu único “sintoma” é a suscetibilidade a reações de Hipertermia Maligna (HM). Por outro lado, a MHS também pode ocorrer em pacientes com Doenças Relacionadas ao RYR1 (RYR-1-RD) com sinais e sintomas típicos de miopatia (fraqueza muscular). Os gatilhos para Hipertermia Maligna incluem certos medicamentos usados para anestesia geral, ou seja, quando alguém é “colocado para dormir”, geralmente antes de uma cirurgia. A anestesia geral é usada em uma ampla variedade de ambientes, incluindo salas de cirurgia, salas de emergência e unidades de terapia intensiva (UTI). Medicamentos específicos conhecidos por desencadear a Hipertermia Maligna incluem anestésicos administrados por via intravenosa, e inalatória via tubo respiratório, como segue:
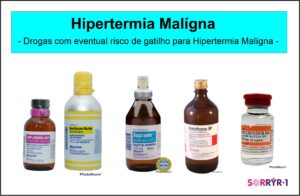
Mutações no RYR1 associadas a Hipertermia Maligna (HM) mostram uma penetrância variável, e isso significa que uma pessoa pode passar por várias exposições a gatilhos sem problemas antes que uma reação de HM ocorra pela primeira vez. Para tornar as coisas ainda mais confusas, as pessoas com a mesma mutação (incluindo membros da mesma família) podem ter reação clínica diferente, o que significa que algumas podem ser sensíveis ao calor, algumas podem ter reações de HM à anestesia, algumas podem ter rabdomiólise com exercícios físicos, e algumas podem não ter problemas com nenhuma dessas condições.
QUADRO CLÍNICO
O quadro clínico de um episódio/crise de Hipertermia Maligna é variável, e compreende manifestações de alterações metabólicas, de lesão muscular, e das complicações secundárias. Esta condição é expressa por rigidez muscular, aumento do consumo de oxigênio e produção de gás carbônico, acidemia (respiratória e metabólica), taquicardia, taquipnéia, hiperpotassemia, rabdomiólise e mioglobinúria. A dessaturação da hemoglobina no sangue arterial pode ser identificada por oximetria de pulso. Entre os diversos fatores que potencialmente contribuem para a dessaturação persistente, encontram-se acidemia, hipercarbia e hipertermia, capazes de deslocar a curva de saturação da hemoglobina para a direita. A hipercarbia, já detectada na cartografia, parece preceder as demais manifestações. A forma fulminante da Hipertermia Maligna é caracterizada por hipercapnia, rigidez muscular, hipertermias graves, e rabdomiólise, mas situações como cirurgias cardíacas sob circulação extracorpórea (CEC) com hipotermia podem atenuar a expressão clínica da Hipertermia Maligna (HM). A hiperventilação pode mascarar o diagnóstico de HM. Bloqueadores neuromusculares podem retardar o início das manifestações da crise de HM. Convém destacar que nem sempre hipertermia é manifestação inicial ou proeminente da HM. A rigidez muscular pode inexistir em 25 % dos casos, e a Hipertermia ser registrada em apenas um terço deles. A HM surge a qualquer momento durante a anestesia, tendo sido descrita sua ocorrência até 3 horas após a interrupção da exposição ao agente desencadeante (gatilho). A crise de Hipertermia Maligna (HM) pode manifestar-se tardiamente, mesmo após a interrupção da administração do agente desencadeante (gatilho), talvez a imobilidade determinada pela própria anestesia limite a liberação de cálcio a partir do retículo sarcoplasmático. Ao acordar, aumenta a atividade muscular e, na presença de resíduos anestésicos, vêm-se potencializadas à liberação intracelular de cálcio e seus efeitos metabólicos. Tem-se a impressão de haver diferenças entre os halogenados com relação ao seu potencial para desencadear crises de Hipertermia Maligna (HM). O halotano parece ser o de maior risco. A exposição ao isoflurano pode associar-se à crise de HM de início tardio. Parece que a indução da liberação de cálcio do retículo sarcoplasmático pelo sevoflurano é menos intensa em comparação aos demais agentes.
Observação de Manifestações Clínicas Iniciais: Taquicardia - 96,0%, Rigidez muscular - 83,6%, Instabilidade hemodinâmica - 85,5%, Taquipnéia - 85,0%, Cianose - 71,1%, Hipertermia - 30,0%
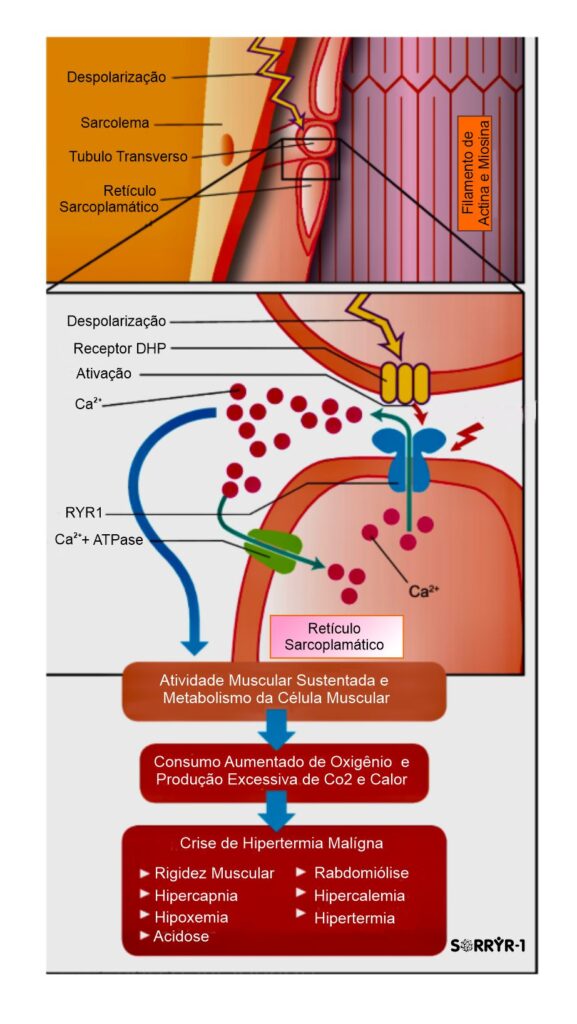
TERAPIA
Dada a gravidade da doença, o tratamento da Hipertermia Maligna no regime operatório deve ser iniciado em caráter de emergência. A administração do anestésico deve ser interrompida imediatamente, e o paciente deve então receber infusão intravenosa de dantroleno sódico, um relaxante muscular que restaura os níveis fisiológicos de cálcio nos músculos.
Posteriormente, o paciente é induzido a baixar a temperatura corporal por meio de fluidos frios e bolsas de gelo para evitar consequências no cérebro, e é administrado oxigênio para satisfazer o aumento da demanda do organismo. Além disso, a acidose metabólica induzida por lactato é prontamente tratada e o desequilíbrio eletrolítico corrigido. O sucesso da intervenção depende em grande parte da rapidez no reconhecimento dos sintomas e da resposta individual do paciente à terapia.
Nos últimos trinta anos, graças a novos estudos e descobertas no campo farmacológico, a taxa de mortalidade da hipertermia maligna caiu drasticamente, de 70-80 % para 5%, tornando-se uma doença relativamente manejável e tratável.

