As doenças musculares são aquelas que afetam a estrutura e funcionamento do músculo, sendo as principais: as distrofias musculares, as miopatias congênitas, as miopatias inflamatórias e as miopatias endócrinas e metabólicas. É importante destacar que cada uma delas possui suas variações que também se diferenciam.
Essas doenças já foram muito confundidas em diagnósticos no passado, e fico triste, porque isso ainda tem acontecido nos dias de hoje, mesmo com os avanços científicos. A única razão que acredito ser ainda a causa para essa confusão nesses diagnósticos seria por essas doenças serem consideradas “doenças raras”, portanto, muitas vezes desconhecidas por parte da comunidade médica. Assim, pode haver a falha no momento dos exames clínicos, ponto inicial para diagnóstico de qualquer doença.
Eu mesmo vivi uma experiência dessa, pois no decorrer de grande parte da minha vida eu recebi vários “diagnósticos” de Distrofia Muscular Congênita (DMC) do tipo: Duchenne, Facioescapuloumeral e Cinturas. E, os prognósticos foram do pior a até o mais brando. Estes diagnósticos ou hipóteses de diagnósticos vieram até de importantes instituições, como de uma clínica indicada pelo MDA (Muscular Distrophy Association), maior referência ligada a essa doença.
Deve-se levar em consideração que, naquela época, pouco se sabia sobre essa doença, nem tão pouco sobre a genética humana; contudo, um erro de diagnóstico hoje em dia seria inaceitável. Essa situação causou em mim grandes transtornos, de emocional aos físicos. Somente aos 44 anos de idade foi que finalmente obtive meu correto e “definitivo diagnóstico”, ou seja, de que sou portador de Miopatia Congênita Centronuclear (MCC), causada pela mutação no gene RYR1.
A Distrofia Muscular Congênita e a Miopatia Congênita Centronuclear apresentam várias características em comum, tais como: são doenças de origem genética, afetam os músculos esqueléticos, caracterizam-se clinicamente por hipotonia e fraqueza muscular, geralmente apresentam-se desde o nascimento, têm curso clínico estático ou lentamente progressivo. Essas doenças não tem cura, e o tratamento envolve terapia de suporte, como fisioterapia, dispositivos de mobilidade e, em alguns casos, medicamentos. Mesmo assim, as duas doenças neuromusculares diferem entre si.
Daí, eu volto com a questão sobre as falhas nos diagnósticos, já que muitos médicos se prendem somente ao resultado do exame genético e não conhecerem os sinais clínicos das diferentes doenças e particularidades dos indivíduos afetados.
Assim, essas noções devem ser levadas em consideração por três razões: Primeiro, muitas das miopatias congênitas podem ser causadas por mutações em mais de um gene, o que sugere um impacto da heterogeneidade genética. Segundo, mutações no mesmo gene podem causar diferentes patologias musculares. Terceiro, a mesma mutação genética pode levar a diferentes características patológicas e sintomatológicas em membros da mesma família ou no mesmo indivíduo em idades diferentes.
Em resumo, eu destacaria que tanto a Distrofia Muscular, quanto a Miopatia Congênita Centronuclear são de origem genética, mas distintas em termos de suas características clínicas e podem variar em gravidade de pessoa para pessoa. Enquanto a Distrofia Muscular envolve a degeneração progressiva dos músculos devido a problemas na estrutura das proteínas musculares, a Miopatia Congênita Centronuclear é caracterizada por uma anormalidade na localização dos núcleos das células musculares. Assim, é importante consultar um médico especialista para um diagnóstico preciso, para que se possa ser feito um acompanhamento adequado do caso, pois o tratamento pode variar dependendo da condição clínica específica de cada indivíduo.
Os portadores de doenças relacionadas ao RYR-1 através do SORRYR-1 comemoram junto com a comunidade dos portadores de Distrofia Muscular, em especial os Duchennes, pela recente aprovação pelo FDA (Federal Drug Administration) do ELEVIDYS (delandistrogene moxeparvovec-rokl) destinada ao tratamento de pacientes pediátricos ambulatorial com idades entre os 4 e os 5 anos com Distrofia Muscular tipo Ducehene #Duchenne #MuscularDystrophy (#DMD) com mutação confirmada no gene DMD.
O SORRYR-1 cumprimenta também a Sarepta Therapeutics @sareptatherapeutics pelo importante trabalho em pesquisas e desenvolvimento de medicamentos genéticos de precisão potencialmente transformadores para doenças raras. O ELEVIDYS é a primeira terapia gênica para DMD (Distrofia Muscular tipo Ducehene) e foi projetada para atingir a causa subjacente da doença.
O SORRYR-1 destaca a crucial importância do MDA (Muscular Dystrophy Association) @mdaorg que esteve presente desde o início nos trabalhos de pesquisas da terapia genética, e criou recentemente uma Rede de Apoio à Terapia Gênica (MDA Gene Therapy Support Network - GTx) para fornecer recursos e orientação para médicos e famílias. O MDA (Muscular Dystrophy Association) é a primeira organização de saúde voluntária nos Estados Unidos voltada para pessoas que vivem com distrofia muscular, e doenças neuromusculares, e que a mais de 70 anos abriu o caminho para acelerar as pesquisas, promoção do atendimento e defesa ao apoio de famílias e indivíduos afetados.
O SORRYR-1 entende esta grande conquista para o tratamento dos portadores de Duchene como uma grande prova dos avanços científicos e empenho da indústria farmacêutica, reforçando assim, a esperança de nós, portadores de doenças relacionadas à mutação no RYR-1, de que uma terapia semelhante possa surgir em um futuro próximo em nosso benefício como tratamento e cura.
TERAPIA GENÉTICA
A terapia genética trata uma doença agindo no próprio gene mutado. A terapia genética pode introduzir um gene para ajudar a combater doenças, substituir um gene mutante, editar um gene mutante ou excluir um gene mutante. Em teoria, as terapias genéticas oferecem o potencial de cura, não apenas de tratamento.
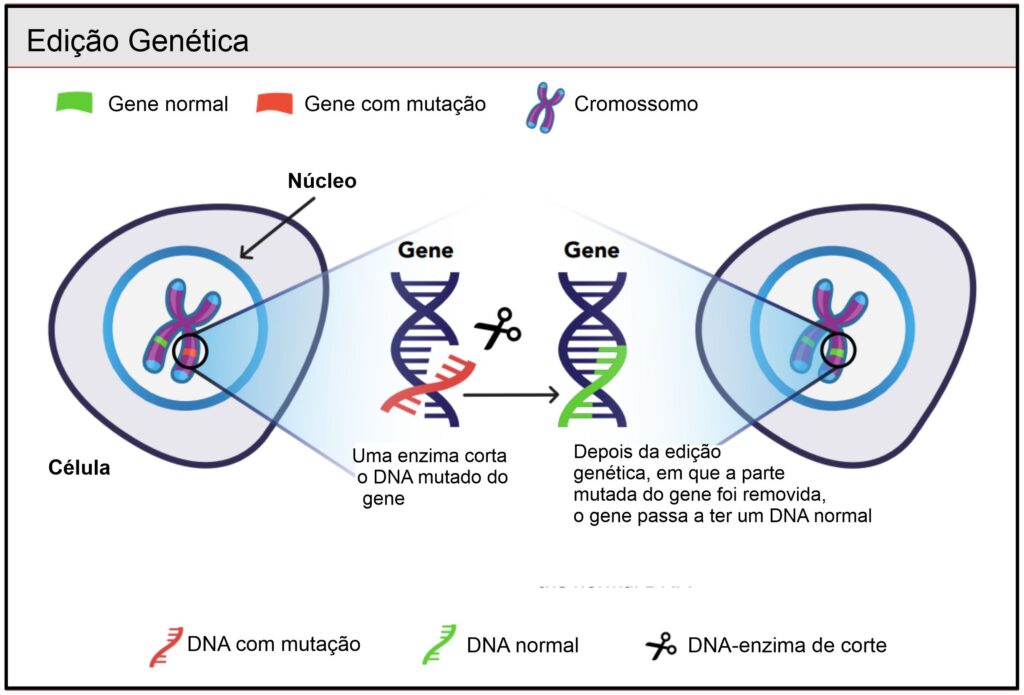
Edição de Gene
Na terapia genética, a edição de genes tem o objetivo de corrigir ou “editar” apenas uma pequena parte do gene, sendo esta, uma abordagem que visa tornar as mudanças mais precisas e permanentes.
RYR-1 Foundation Clinical Care Guidelines
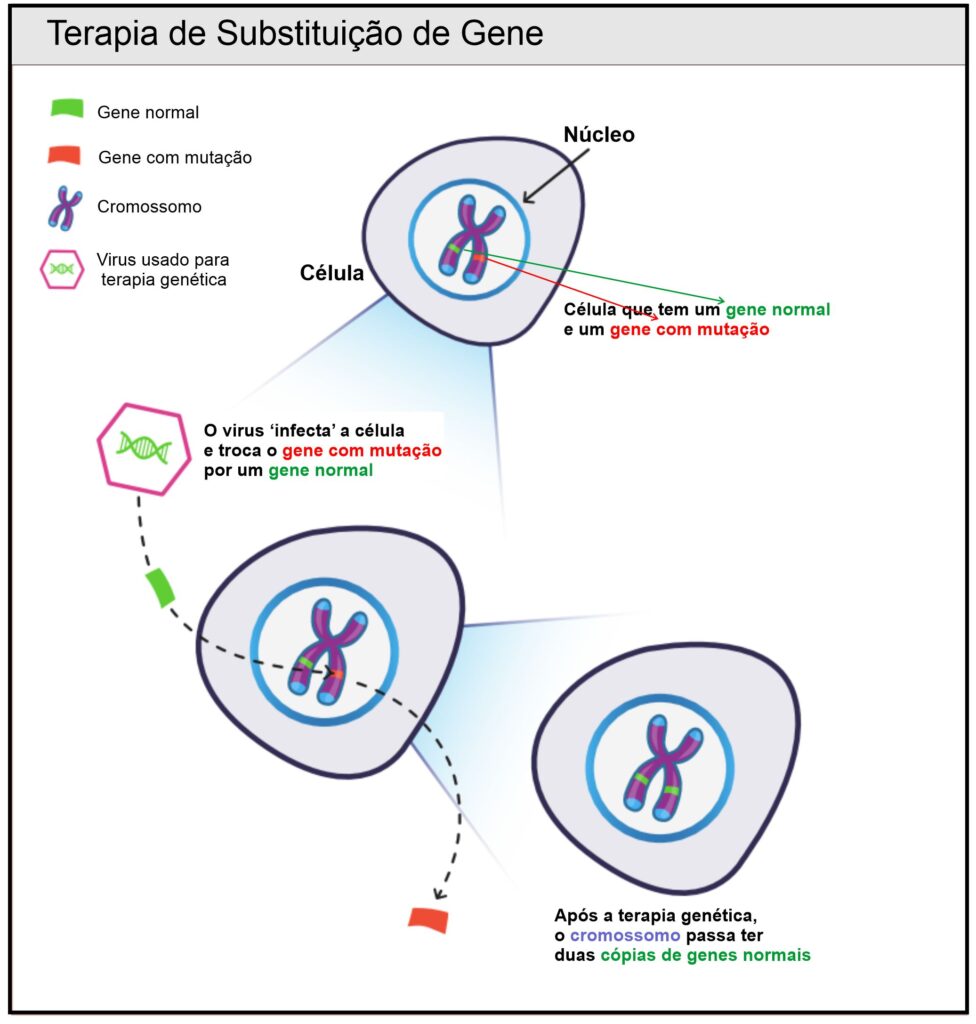
Terapia de Substituição de Gene
A terapia de substituição de gene se dá com a introdução de uma molécula, chamada de vetor, que carrega uma cópia normal de um gene. Os vetores geralmente são vírus, porque podem entrar em uma célula. No entanto, os vírus são projetados para não deixar as pessoas doentes. Alguns vetores comumente usados, chamados Vírus Adenoassociados, levam o gene normal para o núcleo da célula. Ao substituir o gene defeituoso (mutado) por uma nova cópia normal do gene, a célula agora produz uma proteína normal em vez da proteína anormal causadora de doenças.
Atualmente, a terapia de substituição de genes usando um vetor viral padrão não é viável para as doenças relacionadas ao RYR-1, porque o gene RYR1 é muito grande para ser empacotado nos vetores virais comumente usados. A terapia de substituição de genes poderá se tornar uma opção terapêutica no futuro, quando outros vetores paraentrega de genes poderão serem descobertos, ou quando técnicas totalmente novas poderão ser inventadas.
RYR-1 Foundation Clinical Care Guidelines
Lembre-se de que um gene é um segmento de DNA, um código de instruções sobre como produzir proteínas importantes. O DNA é uma molécula de fita dupla muito longa que tem uma forma helicoidal torcida, como uma escada sinuosa. Os blocos de construção do DNA são chamados de nucleotídeos. Existem quatro tipos de nucleotídeos: adenina (A), citosina (C), guanina (G) e timina (T). Uma importante coisa a lembrar é que a sequência de nucleotídeos (A,C,T e G) é a instrução que suas células usam para produzir proteínas. Na maioria dos casos, as doenças genéticas graves são causadas por um único erro ortográfico deste código, por exemplo, você tem um "A" onde deveria haver um "G". Esses erros no código de DNA podem impedir que suas células produzam uma proteína importante ou uma versão ruim disso. O objetivo da edição genética é corrigir permanentemente essas mudanças, alterando a sequência do seu próprio DNA. Pense em levar uma borracha e um lápis para corrigir um erro de ortografia em uma carta manuscrita. Em princípio, isso parece fácil, mas ainda é um grande desafio de fazê-lo de forma correta, eficiente e segura em todas as células.
A maioria das abordagens atuais de edição de genes envolve o uso de proteínas chamadas nucleases as quais cortam o DNA em locais específicos. Em geral, você precisaria de cortar o DNA apenas no local ou próximo ao nucleotídeo incorreto. Pense em colocar a borracha exatamente na palavra que você precisa reescrever. Você não gostaria de cortar DNA em outros lugares, porque isso poderia causar erros perigosos que mudariam o significado do código e fariam com que suas células funcionassem incorretamente.
A mais comumente nuclease usada para edição de genes é o sistema CRISPR/Cas9. Esta técnica é composta por duas partes: Cas9, que é uma proteína que foi descoberta em bactérias, e um RNA Guia. Cas9 e o RNA Guia se unem para formar a nuclease ativa, que você pode imaginar como borracha para a carta manuscrita. Essas tesouras se ligarão ao DNA com base na sequência do RNA Guia. Os cientistas podem alterar a sequência do RNA Guia conforme necessário para cortar quase qualquer sequência de DNA, com algumas limitações. Uma vez que o DNA é cortado, ele será reparado pela célula. Controlar o processo de reparo do DNA determina quais tipos de mudanças podemos fazer ao DNA. Ao contrário da edição precisa em uma carta manuscrita, ainda não temos controle sobre como a sequência será alterada. Alguns tipos de mudanças são fáceis de fazer, enquanto outros são mais difíceis e menos previsíveis.
Além de CRISPR/Cas9 e outras nucleases de edição de genes (por exemplo, dedos de zinco), também existem novas ferramentas promissoras em desenvolvimento. “Editores de base” são versões modificadas do sistema CRISPR/Cas9 que não cortam completamente a molécula de DNA de fita dupla. Em vez disso, eles cortam apenas uma fita, e mudam quimicamente um nucleotídeo para outro dentro de uma determinada janela de edição. Pense como mudar um “l” para um “t” adicionando um traço horizontal a um documento manuscrito. Nenhuma borracha foi necessária, já que esta versão do Cas9 não corta completamente o DNA. Portanto, o risco de falta de palavras é minimizado. A eficácia de editores de base está melhorando e eles têm vantagens significativas para corrigir alterações de nucleotídeo único. No entanto, mais trabalho é necessário para melhorar sua precisão.
A adição mais recente à caixa de ferramentas de edição de genes é o Prime Editing, ou Sistema de Edição Principal. A vantagem de “Prime Editors” é que eles nos deixariam corrigir praticamente qualquer tipo de pequeno erro no DNA, que atualmente não é possível com editores de base. Por exemplo, você pode modificar qualquer letra do alfabeto para qualquer outra, não apenas convertendo “l” para “t” (como no exemplo anterior). No entanto, muito mais trabalho é necessário para demonstrar que eles são seguros, eficazes e confiáveis.
Para resumir, a Edição de Genes é uma tecnologia muito empolgante que só recentemente se tornou possível, principalmente em um ambiente de laboratório de pesquisa. Em contraste com a terapia gênica tradicional, o objetivo é precisamente mudar o DNA do próprio paciente em cada célula do tecido afetado. Isso seria garantir que as mudanças forneçam um conjunto preciso e permanente de instruções de DNA que duraria por toda a vida. Embora esse campo seja uma grande promessa, as ferramentas atuais ainda têm grandes limitações, sendo que um dos maiores é entregar o mecanismo de edição do genoma a intencionada célula alvo do corpo do paciente. Outras considerações incluem o controle de qualquer resposta imune a impedir que o corpo de um paciente ataque as ferramentas de edição de genes. Além disso, maior precisão é necessário para controlar os tipos de alterações a serem feitas no local pretendido (no alvo edição), bem como evitar erros em outras partes do DNA de um paciente (edição fora do alvo). Apesar desses desafios, o futuro da edição de genes é muito brilhante e é algo para se esperar nos próximos anos.

