
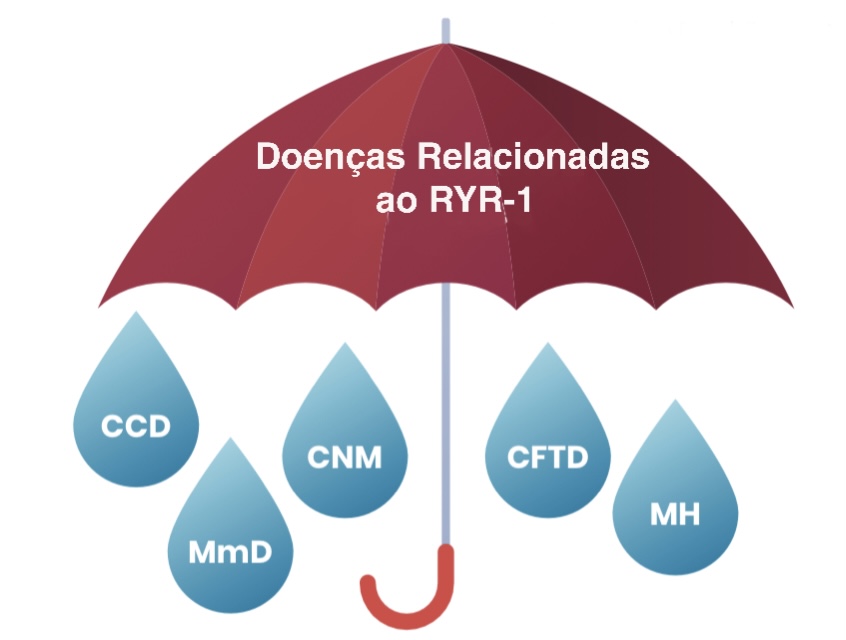
DOENÇAS RELACIONADAS AO RYR-1 É UM TERMO “GUARDA-CHUVA”, OU SEJA, QUE ABRANGE ALGUMAS DIFERENTES DOENÇAS MUSCULARES
As doenças relacionadas ao RYR1 são raras, e classificadas como doenças órfãs , “órfãs”, se trata de um termo usado para identificar uma doença que afeta uma pequena percentagem da população. A verdadeira prevalência dessas doenças é difícil de calcular, pois muitos casos são mal diagnosticados ou não diagnosticados. Também há relatos de prevalência ligeiramente aumentada em certas populações étnicas e geográficas.
As doenças relacionadas ao RYR1 são devidas a uma mutação ou mutações no gene RYR-1. Na prática, podemos sintetizar o processo de mutação da seguinte maneira: o gene RYR-1 codifica o receptor RYR-1, o qual é um canal de cálcio no retículo sarcoplasmático do músculo esquelético; o fluxo de cálcio através do receptor RYR-1 é um componente crítico para a excitação-contração muscular. Uma mutação no gene RYR-1 pode alterar o número, estrutura e/ou função do receptor RYR-1, podendo assim desencadear uma ampla gama de sintomas, e consequências clínicas diferentes, que podemos chamar de doenças relacionadas ao RYR1.
Historicamente, indivíduos com RYR1-RD eram diagnosticados com base em características da biópsia muscular, como núcleos centrais, bastonetes e desproporção do tipo de fibra, embora essas características não sejam exclusivas da RYR1-RD e possam mudar ao longo do tempo. Com o surgimento de mais doenças ligadas a variantes do RYR1 — como a síndrome de King-Denborough, rabdomiólise induzida por exercício e miopatias de início na idade adulta, a sobreposição diagnóstica aumentou. Por isso que para abranger a crescente gama de condições ou doenças relacionadas ao RYR1, incluindo casos de início na idade adulta recém-identificados, foi sugerido o uso do termo “doenças relacionados ao RYR1 (RYR1-RD)” como uma nomenclatura unificada para esse complexo espectro de doenças.
“Doenças Relacionadas ao RYR1 – RYR1-RD” é um termo abrangente que engloba uma gama de subtipos relacionados ao RYR1 que afetam o sistema neuromuscular em humanos. O Orphanet, ( https://www.orpha.net/ ) banco de dados europeu de doenças raras, lista diversas condições relacionadas ao gene RYR1 (figura a seguir):

Alguns pesquisadores classificam as doenças relacionadas ao RYR1 em vários subtipos com base nos seguintes critérios:
Achados de biópsia muscular (histopatologia):
- Miopatia Central Core - Central Core Disease (CCD)
- Doença Multi-Minicore (MmD)
- Miopatia Congênita Centronuclear - Centronuclear Myopathy (CNM)
- Desproporção Congênita do Tipo de Fibra - Congenital Fiber-Type Disproportion (CFTD)
Sintomas (fenótipo clínico):
- Síndrome de King-Denborough (KDS)
- Síndrome de Rabdomiólise
- Miopatia Axial de Início Tardio (LOAM - Late-onset axial myopathy)
Interações fármaco-gene (farmacogenética):
- Suscetibilidade à Hipertermia Maligna (SHM)
- Miopatia Induzida por Estatina
Os sintomas das doenças relacionadas ao RYR1 geralmente estão presentes desde o nascimento (congênitas) ou aparecem na primeira infância e podem ser estáticos, dinâmicos ou uma combinação de ambos. Os sintomas estáticos (presentes o tempo todo) incluem fraqueza muscular, atraso motor, dificuldades para andar e subir escadas, escoliose, fraqueza muscular facial e fraqueza muscular ocular (oftalmoparesia). Os sintomas dinâmicos (que aparecem e desaparecem com base em certos gatilhos) incluem doenças relacionadas ao calor, degradação muscular induzida por exercício (rabdomiólise), dor muscular (mialgia), cãibras musculares e fadiga.
As variantes do RYR1 também são a principal causa de suscetibilidade à hipertermia maligna (SHM), representando >60% dos casos. A hipertermia maligna (HM) é uma reação potencialmente fatal que ocorre em indivíduos suscetíveis após exposição a anestésicos voláteis ou relaxantes musculares despolarizantes, os quais desencadeiam um rápido aumento da temperatura corporal (hipertermia) e degradação muscular (rabdomiólise). As reações de HM são tratadas com o medicamento dantroleno.
Os sintomas apresentados por indivíduos com doenças relacionadas ao gene RYR1 podem ser bastante variáveis; no entanto, o curso da doença geralmente não é progressivo ou é muito lentamente progressivo. A expectativa de vida é geralmente normal em indivíduos afetados e o desenvolvimento cognitivo não é afetado. Embora não haja cura ou tratamento aprovado para doenças relacionadas ao gene RYR1, estratégias de suporte, incluindo fisioterapia, podem ajudar a controlar as limitações funcionais e promover uma alta qualidade de vida.
Como dito anteriormente, as Doenças Relacionadas a RYR-1 (RYR-1-RD) engloba diferentes tipos de doenças relacionados ao RYR1, e é também demonstrado como um termo “guarda-chuva” se referindo às suas subdivisões.
Miopatia Central Core - Central Core Disease (CCD)
Miopatia Central Core - Central Core Disease (CCD)
Descrita pela primeira vez por Magee e Shy em 1956. A DNC geralmente é herdada de forma dominante. Quando observadas ao microscópio, as fibras musculares com DNC apresentam coloração escura, mas também áreas claras no meio das fibras, que não são coradas. Essas áreas claras representam a ausência de atividade mitocondrial. As mitocôndrias são as estruturas responsáveis pela geração de energia para a célula.
A Miopatia Central Core (CCD) causa fraqueza muscular de leve a muito grave; no entanto, a maioria dos indivíduos afetados apresenta fraqueza muscular leve e persistente, que pode piorar com o tempo. Essa fraqueza afeta os músculos próximos ao tronco (músculos proximais), particularmente na parte superior das pernas e nos quadris. A fraqueza muscular também pode fazer com que os bebês afetados pareçam "flácidos" e resultar em atraso no desenvolvimento motor, como sentar, ficar em pé e andar. Bebês gravemente afetados apresentam tônus muscular profundamente fraco (hipotonia), resultando em dificuldades de alimentação e problemas respiratórios graves ou com risco de vida. A CCD também está associada a anormalidades esqueléticas, como curvatura excessiva da coluna vertebral (escoliose), luxação do quadril e deformidades articulares chamadas contraturas, que restringem o movimento de certas articulações. Indivíduos com CCD geralmente conseguem andar durante toda a vida.
Doença Multiminicore (MmD)
Doença Multiminicore (MmD) foi descrita pela primeira vez como doença multicore por Engel e colaboradores em 1971. A MmD é herdada de forma recessiva e causa fraqueza muscular e problemas de saúde relacionados, que variam de leves a potencialmente fatais. Quando observadas ao microscópio, as fibras musculares da MmD apresentam coloração escura, mas também várias áreas claras dentro de cada fibra muscular, sem coloração, resultando em uma aparência "roída por traças". Assim como na CCD, essas áreas sem coloração representam a ausência de atividade mitocondrial. Em geral, a MmD causa sintomas mais graves do que a CCD. Os pesquisadores identificaram quatro formas distintas de MmD:
Forma clássica: a forma mais comum, associada à fraqueza muscular no pescoço (axial) e tronco, com início na infância ou no início da infância, curvatura anormal da coluna vertebral (escoliose), comprometimento respiratório e hiperlaxidão (aumento da flexibilidade) das articulações dos membros.
Forma oftalmoplégica: associada à paralisia ou fraqueza dos músculos oculares, com fraqueza muscular generalizada e fraqueza facial grave.
Forma de início precoce: associada à artrogripose (contraturas articulares desde o nascimento).
Forma lentamente progressiva: associada ao comprometimento dos músculos das mãos.
A fraqueza muscular faz com que os bebês afetados pareçam "flácidos", com tônus muscular fraco (hipotonia), resultando em atraso no desenvolvimento motor, como sentar, ficar em pé e andar. A rigidez da parede torácica e da coluna vertebral também está associada à MmD. Quando combinada com a fraqueza dos músculos necessários para a respiração, podem ocorrer problemas respiratórios graves ou com risco de vida. Quase todas as crianças com MmD desenvolvem uma curvatura anormal da coluna vertebral (escoliose), que surge durante a infância e piora progressivamente com o tempo.
Miopatia Congênita Centronuclear - Centronuclear Myopathy (CNM)
A Miopatia Congênita Centronuclear (CNM) foi descrita pela primeira vez por Spiro e colaboradores em 1966, a MCN é herdada de forma recessiva. Ao serem examinadas ao microscópio, as fibras musculares da CNM apresentam núcleos (estruturas que contêm cromossomos) localizados no centro das fibras musculares, em vez de na periferia. Esse achado na biópsia muscular também foi identificado em diversas outras doenças neuromusculares genéticas, incluindo miopatias relacionadas aos genes MTM1, BIN1 e DNM2.
A Miopatia Congênita Centronuclear (CNM) pode causar fraqueza muscular em qualquer fase da vida, desde o nascimento até o início da idade adulta. Essa fraqueza muscular pode levar a atrasos no desenvolvimento motor (engatinhar ou andar) e pode ser progressivamente lenta. Alguns indivíduos afetados podem necessitar de cadeira de rodas logo na infância. Outros sintomas incluem problemas respiratórios de leves a graves, queda da pálpebra superior (ptose), fraqueza dos músculos faciais, anormalidades nos pés, palato ogival (céu da boca alto) e curvatura anormal da coluna vertebral (escoliose) .
Desproporção Congênita do Tipo de Fibra - Congenital Fiber - Type Disproportion (CFTD)
A Desproporção congênita do tipo de fibra muscular (CFTD) foi escrita pela primeira vez por Brooke e colaboradores em 1969, e é herdada de forma recessiva. Quando observada ao microscópio, o tecido muscular com CFTD apresenta fibras musculares do tipo 1 (contração lenta) que são consistentemente menores do que as fibras musculares do tipo 2 (contração rápida).
A Desproporção congênita do tipo de fibra muscular (CFTD) causa fraqueza muscular, particularmente nos músculos dos ombros, braços, quadris e coxas. A fraqueza também pode afetar os músculos faciais, os músculos extraoculares que controlam o movimento dos olhos (oftalmoplegia) e os músculos da pálpebra superior (ptose). Indivíduos com CFTD geralmente apresentam rosto alongado, palato ogival e dentes apinhados. Os indivíduos afetados podem apresentar deformidades articulares (contraturas) e uma curvatura anormal da região lombar (lordose) ou uma curvatura lateral da coluna vertebral (escoliose). Aproximadamente 30% das pessoas com CFTD apresentam problemas respiratórios leves a graves relacionados à fraqueza dos músculos necessários para a respiração. Algumas pessoas que apresentam esses problemas respiratórios necessitam do uso de um aparelho para auxiliar na regulação da respiração à noite e, ocasionalmente, também durante o dia. Cerca de 30% dos indivíduos afetados têm dificuldade para engolir devido à fraqueza muscular na garganta. Raramente, pessoas com essa condição apresentam enfraquecimento e aumento do músculo cardíaco (cardiomiopatia dilatada). Variantes genéticas causadoras de CFTD também foram identificadas em outros genes, como ACTA1, TPM3 e SELENON.
Suscetibilidade a Hipertermia Maligna - Malignant Hyperthermia Susceptibility (MH)
Indivíduos sensíveis à Hipertermia Maligna (MH) podem experimentar uma vida diária normal sem qualquer sintoma ou fraqueza muscular. No entanto, quando expostos a certos agentes anestésicos, os pacientes podem experimentar um episódio de Hipertermia Maligna. A Hipertermia Maligna caracteriza-se por um estado hipermetabólico, causando um aumento anormal do calor, com rigidez excessiva e quebra muscular associada, e aumento da frequência cardíaca. As complicações graves da HM incluem: lesão cerebral, sangramento interno, parada cardíaca e/ou falência de múltiplos órgãos. As complicações cardiovasculares associadas podem ser fatais. Qualquer indivíduo com uma mutação(ões) do RYR-1, se a anestesia for necessária para um procedimento médico-cirúrgico, é aconselhado a tomar "precauções contra hipertermia maligna". Indivíduos com mutações RYR-1 suscetíveis a MH também correm risco de rabdomiólise, bem como outras dores e cãibras musculares relacionadas ao calor e ao esforço. Rabdomiólise é o termo geral para ruptura muscular associada a uma ampla variedade de gatilhos externos, incluindo: exercícios extenuantes além do limite de fadiga, abuso de drogas ou álcool, uso de suplementos ou certos medicamentos, doença viral recente ou trauma muscular. Os sinais e sintomas da rabdomiólise incluem dor muscular intensa, elevação repentina e queda subseqüente dos níveis séricos de creatina fosfoquinase (CPK) e produtos da quebra muscular na urina ("mioglobinúria"). O curso da rabdomiólise é caracterizado principalmente por mialgia (dor muscular) com aumentos leves a moderados da CPK. Nesses casos leves, muitos indivíduos não procuram atendimento médico. No entanto, em alguns o curso clínico é grave, resultando em hiper-CK-emia profunda, insuficiência renal aguda, síndrome do compartimento, coagulação intravascular disseminada, arritmias cardíacas secundárias a desequilíbrios eletrolíticos e, possivelmente, parada cardíaca se não tratada. Portanto, indivíduos com mutações no RYR-1, especialmente aquelas conhecidas por estarem associadas à suscetibilidade ao HM, devem estar cientes dos gatilhos da rabdomiólise e podem querer consultar um médico antes do início de um regime de exercícios e/ou esportes. Pode haver um papel do dantroleno como agente profilático na prevenção da rabdomiólise e de outros sintomas musculares relacionados ao esforço e ao calor.
