O gene RYR1, descrito em 1989 por MacLennan é responsável por dar as instruções para a proteína do receptor de rianodina 1 (RYR1), que é um canal de cálcio crucial para a contração e relaxamento dos músculos esqueléticos.1 A função principal do gene é codificar o canal que libera íons de cálcio do retículo sarcoplasmático (um reservatório dentro da célula muscular) quando o músculo é ativado por um sinal elétrico. Quando o canal RYR1 se abre, o cálcio entra no citoplasma, permitindo que os músculos se contraiam para permitir o movimento. Variantes (mutações) neste gene podem levar a doenças musculares como miopatias congênitas: Doença do Núcleo Central (Central Core), Doença Multi-Minicore, Miopatia Centronuclear e Desproporção Congênita do Tipo de Fibra. O estudo destas doenças permitiu o sequenciamento do gene RYR1 e a descoberta da relação de variantes neste gene e manifestações clínicas como Hipertermia Maligna (HM), Rabdomiólise e Rabdomiólise por Esforço (RE).
RYR1 está localizado no cromossomo 19q13.1 e codifica o Receptor de Rianodina Tipo 1 (RYR1). O gene RYR1 compreende 106 exons e codifica uma grande proteína com 5038 aminoácidos. Devido ao seu tamanho e complexidade consideráveis, o RYR1 tem sido historicamente um gene desafiador para estudo. Durante muitos anos, os esforços de pesquisa concentraram-se principalmente em três domínios (N-terminal, central e C-terminal) considerados pontos críticos de mutação. No entanto, com o advento das tecnologias de Sequenciamento de Nova Geração (NGS), tornou-se possível analisar o gene inteiro. Como resultado, inúmeras novas variantes foram identificadas fora dos pontos críticos previamente conhecidos. Até o momento, mais de 1000 variantes distintas do gene RYR1 foram relatadas (HGMD; https://www.hgmd.cf.ac.uk/ (acessado em 04 de dezembro de 2025) e LOVD; https://databases.lovd.nl/shared/genes/RYR1 (acessado em 04 de dezembro de 2025), porém apenas 72 foram classificadas como variantes diagnósticas pelo Grupo Europeu de Hipertermia Maligna (EMHG; https://www.emhg.org/diagnostic-mutations (acessado em 04 de dezembro de 2025)). A grande maioria dessas variantes são de sentido trocado, enquanto inserções, deleções e duplicações são relativamente incomuns, representando menos de 10% de todas as variantes patogênicas conhecidas no gene RYR1.
A Hipertermia Maligna (HM) é uma síndrome hipermetabólica farmacogenética que se manifesta como uma crise aguda após a exposição de indivíduos suscetíveis a agentes halogenados e/ou succinilcolina. A crise aguda pode se manifestar clinicamente com hipertermia, hipercapnia, taquipneia, taquicardia, acidose metabólica, hipercalemia, rigidez muscular e rabdomiólise, sendo uma condição extremamente grave. Ocorre em pacientes de todas as etnias e distribuições geográficas, sendo duas vezes mais comum nos homens e menores de 19 anos de idade (50% dos casos). Se não tratada adequadamente, óbito ocorre em 80%–90% dos pacientes. O tratamento consiste em evitar as crises e abordagem seguindo protocolos rígidos nas crises agudas, incluindo-se uso de dantrolene de sódio (reduz a liberação de cálcio do retículo sarcoplasmático do músculo estriado esquelético por limitar a ativação do receptor de rianodina RYR1).2,3
A prevalência da crise de HM é variável, de 1:10.000 em crianças a 1:50.000 em adultos. A frequência relacionada a procedimentos anestésicos varia de 1 em 10.000 a 1 em 250.000, dependendo da população e dos critérios diagnósticos. A suscetibilidade à HM tem sido associada a genes ligados ao metabolismo do cálcio e que codificam proteínas do complexo de acoplamento excitação-contração do músculo esquelético, com uma frequência de 1:217–1:2750 na população geral. O principal gene implicado na suscetibilidade à HM é o RYR1, responsável por aproximadamente 75% dos casos geneticamente confirmados. Famílias raras apresentam variantes nos genes CACNA1S (1%), STAC3 (<1%) ou ASPH.
A apresentação clínica entre portadores de variantes do RYR1 é altamente variável, e o panorama genético é marcado por alta heterogeneidade alélica, penetrância incompleta e expressividade variável. Variantes patogênicas nesse gene resultam em liberação desregulada de cálcio do retículo sarcoplasmático, levando à contração muscular sustentada e crise metabólica.
Devido à heterogeneidade genética e à possibilidade de herança poligênica, o padrão ouro no diagnóstico de suscetibilidade à HM é o teste de contratilidade in vitro fenotípica (IVCT) para o grupo europeu ou o teste de contratilidade com cafeína-halotano (CHCT) para o grupo norte-americano. Atualmente, apenas um teste de contratilidade negativo pode excluir a suscetibilidade à HM, enquanto um teste genético negativo não o faz. No entanto, esforços têm sido feitos para melhorar a capacidade de detecção do teste molecular, que poderia ser uma ferramenta diagnóstica menos invasiva. Por outro lado, variantes no gene RYR1 são muito frequentes na população, e a identificação e classificação dessas variantes como patogênicas requerem testes adicionais e curadoria. Apenas 72 variantes patogênicas ou provavelmente patogênicas no RYR1 são reconhecidas de acordo com a lista do Grupo Europeu de Hipertermia Maligna (EMHG) ( https://www.emhg.org/genetic-scoring-matrix ; acessado em 18 de agosto de 2025), e, além dos critérios do EMHG, existem os critérios do painel de especialistas em curadoria de variantes do ClinGen MHS (VCEP). Variantes missense são as mais comuns, enquanto inserções e duplicações representam menos de 10%. Os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína.
Cada país ou região apresenta diferenças no tipo e na frequência de variantes associadas à HM. A frequência de variantes relatadas em pacientes com HM varia de 37% a 87,5%, sendo as variantes RYR1 p.R614 e p.G2434R as mais frequentes. No entanto, não existem dados genéticos populacionais da América do Sul, exceto pela descrição de casos isolados e famílias. Em estudo recente, desenvolvido na Unifesp – Escola Paulista de Medicina, foram revisados os dados clínicos e laboratoriais de todas as famílias encaminhadas para avaliação na Unidade Brasileira de HM devido a histórico pessoal ou familiar de HM durante anestesia. Foram coletados dados demográficos e clínicos, bem como níveis séricos de creatina quinase (CK), resultados do teste de contratilidade in vitro (TCIV) e resultados de estudos anatomopatológicos do músculo esquelético. A análise molecular foi realizada por meio de sequenciamento de exoma completo (NGS). Pacientes com e sem variantes foram comparados. Variantes no gene RYR1 foram encontradas em 38 pacientes (62,2%), e nenhuma variante foi identificada em 20 pacientes (32,7%). Mais de uma variante no RYR1 foi encontrada em seis indivíduos. Variantes no gene CACNA1S foram encontradas em três pacientes (4,9%), todos com variantes concomitantes no RYR1. Três pacientes apresentaram variantes no gene STAC3 (4,9%). Comparando os grupos de pacientes com variantes no RYR1 com o grupo sem variantes nesse gene, observou-se que o primeiro grupo apresentou valores séricos mais elevados de CK, maior frequência de ptose, estrabismo, e maior amplitude de contratura no TCIV após a administração de cafeína ou halotano. Nesta avaliação preliminar de indivíduos brasileiros com histórico de hipertermia maligna, a frequência de variantes no RYR1 foi semelhante à de relatos anteriores em outros países, porém houve maior frequência de variantes nos genes STAC3 e CACNA1S.4
Também em estudo da Unifesp, foi identificada uma variante rara: duplicação no gene RYR1 na variabilidade do fenótipo de susceptibilidade à Hipertermia Maligna, em uma família com dois irmãos afetados portadores de uma inserção de 18 pares de bases no éxon 91 do gene RYR1, resultando em uma duplicação em fase de 6 aminoácidos (c.12835_12852 dupGAGGGCGCGGCGGGGCTC: 162 p.G4279_T4284insAAGLEG). A expressão relativa do mRNA do gene RYR1 no músculo dos dois pacientes identificou uma redução de aproximadamente 50%, sugerindo um possível alelo hipomórfico. Este achado levanta a questão que os mecanismos patogênicos das variantes do gene RYR1 na Hipertermia Maligna estão principalmente associados a mecanismos de ganho de função, mas pequenas inserções podem frequentemente levar à perda de função ou ao dobramento inadequado da proteína. Este estudo reforça a possibilidade de que a duplicação nessa região possa causar defeitos estruturais e um fenótipo mais grave nos pacientes.5
A Rabdomiólise é uma condição potencialmente fatal que envolve a rápida dissolução do músculo esquelético em resposta a uma variedade de fatores desencadeantes, clinicamente caracterizada por um aumento súbito e acentuado, seguido de uma queda nos valores séricos de creatina quinase (CK) As causas mais comuns de rabdomiólise são lesões por esmagamento secundárias a traumas, esforço físico extremo e miopatias metabólicas. As principais características incluem dor muscular e uma elevação súbita e transitória dos valores séricos de CK. A rabdomiólise grave é frequentemente acompanhada por aumento da excreção urinária de mioglobina (mioglobinúria), o que pode levar à insuficiência renal aguda (IRA) e a uma crise metabólica potencialmente fatal. A ampla gama de complicações (por exemplo, insuficiência renal aguda, arritmias cardíacas, síndrome compartimental, coagulação intravascular disseminada) enfatiza a relevância clínica da rabdomiólise em diversas especialidades médicas.
Estudos de coorte retrospectivos focados em rabdomiólise em pacientes hospitalizados na era pré-sequenciamento de nova geração (NGS) concentraram-se particularmente em fatores desencadeantes externos como a principal causa de um evento de rabdomiólise. Esses estudos identificaram toxinas exógenas (drogas ilícitas, álcool), trauma muscular direto, infecções e exercícios extenuantes (Rabdomiólise por Esforço) como alguns dos fatores desencadeantes mais comuns. Exames genéticos de última geração têm associado mais de 30 genes a uma maior suscetibilidade à rabdomiólise. Contudo, um desafio fundamental na abordagem diagnóstica reside na consideração de quais pacientes necessitam de triagem genética diagnóstica após um episódio de rabdomiólise para identificar uma doença neuromuscular ou metabólica pauci-sintomática ou assintomática. Dentre os genes relacionados, destaca-se o RYR1 e variantes nele podem ser responsáveis por uma proporção substancial de pacientes que apresentam sintomas inexplicáveis de rabdomiólise e/ou mialgia por esforço.6
Com o intuito de revisar a abordagem diagnóstica genética da rabdomiólise, uma pesquisa na Holanda foi realizada, tendo como palavra chave o acrônimo 'RHABDO': Recurrent episodes (episódios recorrentes); HyperCKaemia persisting 8 weeks after the event (aumento de CK persistente após 8 semanas do evento); Accustomed exercise—the intensity of the exercise cannot sufficiently explain the rhabdomyolysis event (exercício de costume – a intensidade do exercício não explica por si o evento de rabdomiólise); Blood CK > 50× the upper limit of normal (ULN) or >10,000 IU/L in female Caucasian patients (aumento de CK > 50x ou > 10 000 UI/L em mulheres); Drugs/medication and other exogenous triggers are insufficient to explain the event (drogas/medicações e outros agentes exógenos são insuficientes para explicarem o evento); and Other affected family members or other exertional symptoms (e.g., severe muscle cramps or swelling) (outros familiares afetados ou com sintomas relacionados ao exercício físico). O acrônimo foi baseado em uma revisão da literatura e em nossa experiência clínica e, portanto, atingiu o nível de evidência de opinião de especialistas. A relevância do RHABDO é ainda mais reforçada por um recente workshop do Centro Neuromuscular Europeu (ENMC) envolvendo 21 médicos e pesquisadores de 12 países diferentes, que enfatizou a necessidade de pesquisa coordenada nesta área. Neste estudo retrospectivo bicêntrico, 122 pacientes foram incluídos. Os fatores desencadeantes mais frequentemente relatados que contribuíram para eventos de rabdomiólise foram exercício (72%), febre/infecção (22%) e/ou medicação (18%). Eles foram submetidos a avaliação genética através de painéis genéticos relacionados à miopatia metabólica (82%), sequenciamento de Sanger (49%) e sequenciamento de exoma completo (NGS) (24%), dos quais 52 pacientes (43%) foram submetidos a múltiplos métodos. Uma variante (provavelmente) patogênica foi identificada em 13 pacientes (11%), todos com ≥2 características de RHABDO presentes. O valor preditivo positivo para ≥2 características foi de 14%, enquanto o valor preditivo negativo foi de 100%. Variantes no gene RYR1 foram descritas em quatro pacientes, um relacionado com esforço (RE).7
A Rabdomiólise por Esforço (RE) é uma degradação muscular patológica associada à atividade física extenuante e agravada por múltiplos fatores de risco. Estes incluem baixo nível de condicionamento físico, alto índice de massa corporal, infecção viral em curso e altitude e temperatura elevadas. A sua incidência é de aproximadamente 36,5 por 100.000 pacientes-ano em atletas ou uma taxa semelhante em militares. De modo geral, as diferenças fundamentais entre a RE e outras formas de rabdomiólise residem em suas causas, fatores desencadeantes e populações afetadas. A patologia celular da lesão por esforço repetitivo (LER) centra-se na ruptura da integridade das células musculares, particularmente no que diz respeito à homeostase iônica e à produção de energia. Exercícios extenuantes ou incomuns podem causar lesão direta ao sarcolema e/ou levar à falha na produção de energia, comprometendo a função de bombas iônicas essenciais, como a Na + /K + -ATPase e a Ca2 + -ATPase. Esse comprometimento aumenta a permeabilidade celular aos íons sódio, resultando em um influxo significativo de cálcio (Ca2 +) para as fibras musculares. O aumento da concentração intracelular de cálcio ativa enzimas dependentes de cálcio, incluindo proteases e fosfolipases, que iniciam a destruição de proteínas miofibrilares, citoesqueléticas e de membrana. Esse processo leva à necrose das fibras musculares, liberando conteúdos intracelulares como CK, mioglobina e eletrólitos no fluido extracelular e na circulação sanguínea. O ciclo vicioso resultante envolve contração muscular sustentada devido ao aumento do cálcio, o que esgota ainda mais as reservas de energia e exacerba o dano muscular.
Embora a via celular geral envolvendo sobrecarga de cálcio e depleção de energia, seja compreendida como o mecanismo de dano às células musculares, os mecanismos subjacentes específicos da rabdomiólise (RB) não são universalmente compreendidos. Estudos em modelos animais, como cavalos suscetíveis à RB recorrente, utilizaram com sucesso a análise do transcriptoma (RNA-seq ou análise de microarray) para revelar alterações na expressão gênica em vias relacionadas à regulação do cálcio, estresse oxidativo e função mitocondrial, demonstrando que essas alterações moleculares podem persistir mesmo entre os episódios. No entanto, as fontes fornecidas não oferecem uma visão abrangente de pesquisas semelhantes em larga escala sobre o transcriptoma, conduzidas especificamente em populações humanas com RB. Um estudo elegante com sequenciamento de RNA em amostras de músculo esquelético de 19 pacientes humanos com histórico de RE, coletadas no mínimo seis meses após o evento de RE mais recente, e oito controles saudáveis para investigar o perfil transcriptômico da RE revelou uma forte supressão da função mitocondrial. Essa supressão incluiu as vias da “cadeia de transporte aeróbico de elétrons” e da “fosforilação oxidativa”, indicando comprometimento da produção de energia. Por outro lado, houve uma regulação positiva de genes associados à adesão e às vias relacionadas à matriz extracelular (aumento do desenvolvimento da matriz extracelular), indicando restauração ativa da função muscular em casos de RE meses após o evento agudo.8
Em síntese, destacamos o que estas pesquisas nos ensinam:
Como o gene RYR1 afeta a função muscular
Associação de variantes (mutações) no RYR1 e Hipertermia Maligna (HM)
Como o defeito no RYR1 leva à Rabdomiólise
O que acontece durante a Rabdomiólise por Esforço (RE)
O que os indivíduos com variantes no RYR1 precisam saber
Hipótese
Referências Bibliográficas:
A Miopatia Congênita Centronuclear é uma doença que causa muitas exclamações, mas também, interrogações. Sempre que as pessoas chegam até mim para questionar sobre minha doença, elas fazem uma exclamação dizendo, “nunca tinha ouvido sobre sua doença !”, e em seguida questionam como ela me afeta, e mais uma vez elas exclamam dizendo, "puxa vida !", e por ultimo exclamam novamente, “essa é uma super rara !”, daí respondo, "sim, no pé da palavra, ela é uma Doença Rara ou talvez uma Doença Ultrarrara.
A Miopatia Congênita Centronuclear decorrente de mutações no gene RYR1 é classificada como Doenças Raras ou Doenças Ultrarraras ?
Entender essas condições é essencial para oferecer o suporte adequado e assegurar abordagens terapêuticas personalizadas tanto para o paciente quanto para seus familiares. Doenças raras nem sempre recebem a atenção que merecem porque afetam relativamente poucas pessoas. Muitas vezes, pode levar anos para que uma pessoa receba o diagnóstico correto de uma doença rara, e cerca de 95% das doenças raras e ultrarraras ainda não têm tratamento.
Diferentes partes do mundo, as doenças raras e ultrarraras são identificadas e tratadas de maneiras diferentes, mas de um modo genérico a definição é mais ou menos assim:
Uma Doença Rara é aquela que acomete no máximo 65 pessoas a cada 100.000 habitantes, o que corresponde a uma prevalência aproximada de 1 para cada 1.500 indivíduos. A maior parte dessas condições tem origem genética, embora algumas possam surgir devido a fatores infecciosos ou relacionados ao sistema imunológico.
A Doença Ultrarrara, como o próprio nome indica, corresponde a condições ainda mais incomuns, com uma incidência de cerca de 1 caso para cada 50.000 indivíduos. Por serem extremamente infrequentes e pouco conhecidas, essas doenças geralmente apresentam maior dificuldade diagnóstica e exigem cuidados altamente especializados e personalizados.
Com relação à questão levantada no início do texto, a prevalência exata da Miopatia Congênita Centronuclear causada especificamente pela mutação no gene RYR-1 é desconhecida.
A dificuldade em determinar um número exato ocorre porque:
• Ela é uma doença ultrarrara por estar dentro de um grupo de doenças já raras, as Miopatias Congênitas.
• Os dados epidemiológicos costumam ser apresentados para a Miopatia Congênita Centronuclear (MCCN) considerando o conjunto das patologias que compõem esse grupo, que inclui mutações nos genes MTM1, DNM2, BIN1 e RYR1.
• O gene RYR1 está associado a um espectro de doenças musculares conhecidas como Doenças Relacionadas ao RYR1 ou RYR1-DR, sendo a forma Centronuclear apenas uma das possíveis manifestações, juntamente com a Miopatia Central Core (MCC), e outras.
Embora não haja um número específico de prevalência para a Miopatia Congênita Centronuclear causada pela mutação no RYR1, podemos contextualizar com base nas informações disponíveis para as condições relacionadas:
• No grupo da Miopatia Congênita Centronuclear (MCCN) em geral, a prevalência geral do grupo é desconhecida. A forma mais comum dentro deste grupo é a Miopatia Miotubular (ligada ao X) causada por mutações no gene MTM1, cuja incidência é estimada em cerca de 1 em 50.000 indivíduos do sexo masculino.
• No grupo das Doenças Relacionadas ao RYR1 (RYR1-DR) em geral, a doença mais frequentemente associado é a Miopatia Central Core (MCC), que é a forma mais comum de miopatia congênita não distrófica.
Um estudo de uma série pediátrica na Espanha estimou a incidência de Miopatias Relacionadas ao RYR1, que abrange a Centronuclear e a Central Core, se mostra em cerca de 1 em 10.000 nascidos vivos na área de estudo.
Em síntese, a mutação no gene RYR1 está associada a um fenótipo de ocorrência rara. A prevalência específica do subtipo Centronuclear (MCCN) ainda não foi estabelecida de forma isolada na literatura médica, sendo considerada extremamente baixa. Entretanto, não há consenso atual sobre classificá-la como no grupo de Doenças Raras ou no grupo de Doenças Ultrarraras.
Na última postagem entitulada, A Importância das Intervenções No Estilo de Vida Para Controle Dos Sintomas da Miopatia Congênita Centronuclear, fiz uma abordagem sobre a importância das condutas multidisciplinar e individualizada, focada na reabilitação e no suporte contínuo para otimizar a funcionalidade e o bem-estar. Esse estilo de vida, seria sustentado por um tripé de condutas, sintetizado no Controle de Stress, Nutrição, e Atividade Física.
Seguindo essa linha de entendimento, gostaria de compartilhar com o leitor sobre minha experiência pessoal, enquanto portador de Miopatia Congênita Centronuclear causada pela mutação genética no RYR-1 com herança autossômica recessiva, com o acompanhamento nutricional que faço com a Dra Victoria Pitombo. Como tanto, segue as gentis palavras da Dra Victoria sobre nossa relação enquanto paciente, e logo a seguir você verá um estudo, base do trabalho aplicado no meu programa nutricional e de suplementação.
" Desde o início do nosso acompanhamento, o que mais me marca no Orlando é o comprometimento real que ele tem com a própria saúde. A nossa relação vai muito além de prescrições: é uma parceria construída com confiança, escuta e respeito por um corpo que exige atenção minuciosa devido à Miopatia Congênita Centronuclear. Cada ajuste que eu faço, desde a ingestão proteica até orientações de digestão e hidratação, tem um propósito maior: proteger sua musculatura, preservar funcionalidade, reduzir estresse oxidativo e garantir mais autonomia e qualidade de vida para ele.
Ao longo desses meses, construímos uma nutrição precisa e totalmente personalizada: calorias calibradas para preservar massa muscular e reduzir gordura abdominal, além de suplementos escolhidos para otimizar função neuromuscular sem gerar desconforto gastrointestinal. Tudo específico para o Orlando. Ver a evolução dele, melhora da autoestima, engajamento e confiança no processo (que ainda temos muito pela frente!) é uma das partes mais gratificantes do nosso trabalho juntos. " Dra Victoria Pitombo
NOTA IMPORTANTE: O estudo apresentado a seguir deve ser entendido como elemento de base científica, portanto, o programa de nutrição e suplementação para cada indivíduo deve ser altamente individualizado e baseado na avaliação clínica, estado nutricional atual e exames laboratoriais. Para garantir segurança e eficácia, as intervenções nutricionais e a suplementação devem ser sempre orientadas e conduzidas por um médico e um nutricionista especializados ou com conhecimento em doenças neuromusculares.
Dra. Victoria C. Pitombo - CRN 58214
℘ Nutricionista especializada a nível de Residência em doenças crônicas pelo Hospital do Coração (Hcor)
℘ Nutricionista Funciona(VP), Esportiva e com especialidade em Obesidade (USP)
Nutrição e Suplementação na Miopatia Congênita Centronuclear Associada à Mutação no Gene RYR1: Evidências Atuais e Considerações Clínicas
As miopatias relacionadas ao gene RYR1 englobam o grupo mais prevalente de miopatias congênitas, incluindo fenótipos como miopatia centronuclear. A mutação compromete a função do receptor rianodina, responsável pela liberação controlada de cálcio intracelular durante a contração do músculo. Como resultado, observam-se fraqueza de predomínio proximal, fadiga rápida, intolerância ao exercício, risco maior de hipertermia maligna e, em muitos casos, comprometimento respiratório.
Embora ainda não exista uma cura, percebe-se consenso crescente de que intervenções nutricionais e suplementação direcionada desempenham um papel importante no tratamento das miopatias. Isso inclui uma melhora na preservação da função muscular, na proteção óssea e na modulação do estresse oxidativo, todos aspectos centrais na fisiopatologia dessa condição.
1. Fundamentos do manejo nutricional
Adequação energética e composição corporal
Pacientes com miopatias congênitas apresentam gasto energético variável, podendo evoluir com desnutrição ou excesso de peso. Ambos são prejudiciais à função muscular e respiratória. Assim, a ingestão energética deve ser individualizada, sempre com foco na preservação da massa magra.
Proteína e estímulo anabólico
A literatura mostra que em doenças neuromusculares a ingestão proteica deve ser 1,2–1,5 g/kg/dia, distribuída ao longo do dia. Para alguns pacientes, pode-se extrapolar a ingestão para até 2g/kg/dia.
A distribuição da proteína ao longo do dia (em no mínimo 4 refeições), otimiza a síntese proteica muscular e reduz períodos de catabolismo, especialmente importante em indivíduos com fadiga crônica e menor resposta anabólica.
2. Suplementação baseada em plausibilidade fisiológica e evidência
Creatina Monohidratada
A creatina possui o potencial de aumentar a disponibilidade de fosfocreatina e melhora a ressíntese de ATP, mecanismo útil em condições que cursam com fadiga muscular rápida. Ensaios clínicos em outras doenças neuromusculares demonstram melhora modesta, mas consistente, na força e na resistência.
Dose usual: Em pacientes com Miopatia congênita centronuclear, o ideal é calcular a dose por kg de peso. Exemplo: Se o paciente pesa 67kg ele deve consumir 67kg x0,1 = 6.7g de creatina por dia.
Leucina
A leucina ativa a via mTOR, estimulando a síntese proteica muscular que é um processo frequentemente diminuído em miopatias com limitação funcional.
Na maioria das vezes, só o suporte de leucina via alimentos não é suficiente, então é interessante suplementar. Dose utilizada em estudos: 2–3 g junto com as refeições principais 2 a 3x por dia, dependendo do paciente. Ou, o paciente pode consumir a leucina 1x ao dia no período noturno.
N-acetilcisteína (NAC)
O estresse oxidativo é um dos mecanismos envolvidos na disfunção do RYR1. A NAC é precursora de glutationa e moduladora do sistema redox. Ensaios clínicos mostram segurança, embora os efeitos em marcadores oxidativos sejam variáveis.
Dose usual: 600–1200 mg/dia.
Outros antioxidantes relevantes
O desequilíbrio redox presente em mutações do RYR1 motiva o uso de antioxidantes adjuvantes:
Vitamina B12
A deficiência de B12 agrava fadiga, fraqueza e neuropatia, podendo intensificar limitações motoras já presentes.
Indicação: quando há deficiência documentada ou fatores de risco.
Doses usuais: 500–1000 mcg/dia (oral) ou protocolo IM médico.
Vitamina D e Cálcio - papel central para músculo e osso
Vitamina D
Há maior prevalência de deficiência de vitamina D em doenças neuromusculares, associada a redução da força, pior função respiratória e menor densidade óssea.
A suplementação e doses deve ser guiada por dosagem sérica (25-OH).
Cálcio
Por que o cálcio é tão importante nesses pacientes ?
O cálcio é o íon mais importante da contração muscular e justamente o elemento cuja movimentação é regulada através do receptor rianodina (RYR1). Em pacientes com mutações nesse gene, ocorrem algumas alterações no fluxo de Ca²⁺, como liberação excessiva ou dificuldade de recaptura, contribuindo para: fadiga precoce;maior risco de dano estrutural das fibras; dificuldade de relaxamento muscular e piora de sintomas ao esforço.
Além disso, muitos pacientes apresentam baixa densidade mineral óssea devido à imobilidade, baixa massa muscular, ingestão reduzida ou pouca exposição solar. Assim, o cálcio torna-se essencial tanto para a função muscular quanto para a prevenção de osteopenia e fraturas, que impactam diretamente mobilidade e autonomia.
Recomendações gerais:
L-Tirosina
A L-tirosina é precursora de catecolaminas e pode modular aspectos de vigília, foco e tolerância ao esforço, elementos frequentemente prejudicados em miopatias crônicas. Estudos sugerem benefício potencial em quadros de fadiga central.
Dose usada em protocolos clínicos: 500–2000 mg/dia.
Considerações finais
Embora a miopatia centronuclear por mutação no RYR1 não possua ainda um tratamento curativo, o manejo nutricional adequado e a suplementação estratégica apresentam papel importante no suporte à função muscular, no controle do estresse oxidativo, na proteção óssea e na melhoria da capacidade funcional diária.
Intervenções como adequação proteica, creatina, leucina, vitamina D e cálcio, NAC, antioxidantes, vitamina B12 e L-tirosina possuem fundamentos fisiológicos consistentes e vêm sendo incorporadas progressivamente à prática clínica, sempre de forma individualizada e integrada ao acompanhamento multiprofissional.
O estilo de vida levado por um indivíduo afetado por uma miopatia congênita é fundamental para o controle dos sintomas da condição, embora não existam tratamentos específicos para a cura da doença. Essas intervenções são consideradas tratamentos de suporte e visam preservar a função muscular, maximizar a independência, e melhorar a qualidade de vida do paciente.
O tratamento da miopatia congênita deve ter uma abordagem multidisciplinar e individualizada, focada na reabilitação e no suporte contínuo para otimizar a funcionalidade e o bem-estar. Esse estilo de vida a que me refiro, seria sustentado por um tripé de condutas, sintetizado no Controle de Stress, Nutrição, e Atividade Física.
 Controle do Stress
Controle do Stress
O controle do stress (stress management) é de extrema importância para a manutenção da saúde e qualidade de vida de indivíduos portadores de uma miopatia congênita. Embora não haja a cura ou tratamento específico para a doença em si, muitas vezes a única terapia indicada é de suporte, como fisioterapia.
Contudo, existem outras condutas que podem ser tomadas visando a mitigação dos sintomas da doença, melhoria na qualidade de vida dos indivíduos, e que inclusive interferem positivamente no prognóstico da doença. Essa conduta diz respeito a uma série de mudança ou incorporação de hábitos vida, chamada de Controle de Stress, e que é composta em vários fatores interligados:
 Nutrição
Nutrição
A qualidade de vida de um indivíduo afetado por uma miopatia é fortemente influenciada pela capacidade de manter a funcionalidade e a autonomia o máximo possível, apesar da fraqueza muscular, que é o sintoma mais comum. Neste contexto, a importância do controle nutricional é crítica e multifatorial, sendo uma parte essencial para seu tratamento de suporte.
No caso específico da Miopatia Centronuclear, por ser uma doença neuromuscular que causa fraqueza muscular significativa (hipotonia), pode levar a várias complicações que exigem um acompanhamento nutricional individualizado e rigoroso, fundamental para minimizar a perda de massa muscular, garantir o aporte energético adequado, e prevenir complicações secundárias à fraqueza. Esse manejo nutricional não visa curar a doença, mas sim otimizar a saúde geral, apoiar a função muscular e respiratória, e prevenir complicações que impactam severamente a qualidade de vida.
Não existe uma dieta ou protocolo de suplementação único para todos os indivíduos com miopatia congênita. A dieta e suplementação deve garantir um aporte energético suficiente para as necessidades metabólicas e para preservar a já tão sofrida massa muscular. O cálculo do gasto energético total precisa ser individualizado, considerando o nível de atividade física e a gravidade da miopatia.
O plano deve ser altamente individualizado e baseado na avaliação clínica, estado nutricional atual e exames laboratoriais. Para garantir segurança e eficácia, as intervenções nutricionais e a suplementação devem ser sempre orientadas por um médico e um nutricionista especializados ou com conhecimento em doenças neuromusculares. Esse planejamento nutricional se baseia em algumas premissas básicas:
Em resumo, uma nutrição bem planejada e a suplementação direcionada são ferramentas de suporte poderosas que visam otimizar a função muscular residual, prevenir deficiências nutricionais, apoiar a saúde óssea e melhorar a qualidade de vida do indivíduo com miopatia congênita.
 Atividade Física
Atividade Física
Sempre digo que a atividade física, no contexto das miopatias congênitas, vai além da função de mero suporte clínico, pois ela se estabelece como um elemento fundamental na promoção da qualidade de vida. A atividade física é crucial não apenas para retardar o declínio funcional, mas também para promover o bem-estar psicossocial do indivíduo afetado pela doença.
Para indivíduos com miopatia congênita, a atividade física é uma ferramenta terapêutica que busca promover a capacidade do indivíduo agir de forma mais autônoma possível, além de atuar na conexão social e dignidade pessoal, situações estas tão importantes quanto qualquer intervenção médica para alcançar uma qualidade de vida.
A atividade física capacita o indivíduo, dando-lhe um sentimento de propósito, controle e autoestima, que são um pilares da qualidade de vida.
A Declaração de Consenso sobre Padrão de Cuidados para Miopatias Congênitas, publicada em 2012 no Journal of Child Neurology, tratou a atividade física no contexto de se enfatizar a importância de um programa estruturado, mas altamente individualizado, para manter a função e a mobilidade.
O consenso reconheceu que, embora não exista cura, a fisioterapia e a terapia ocupacional (TO) são cruciais para o manejo da doença, e como promoção da qualidade de vida do indivíduo.
As diretrizes do consenso de 2012 sobre atividade física e reabilitação se referiam principalmente aos seguintes pontos:
Em essência, a declaração de consenso enfatizou que a atividade física para miopatias congênitas não é uma atividade de "ganho de força" típica, mas sim uma intervenção de manutenção e reabilitação focada em preservar a função e evitar as complicações secundárias da fraqueza, mas sobretudo e principalmente como agente de promoção de qualidade de vida.
Com os recentes avanços da ciência genética, a grande maioria dos diagnósticos de uma miopatia causada pela mutação no gene RYR1 acontece nos primeiros anos de vida até a adolescência. Entendo que foi pensando nesse público, que a Fundação RYR-1 @theryr1foundation resolveu criar um elemento que de alguma forma comunicasse com esse público infanto-juvenil, o qual é uma parte considerável da comunidade RYR1.
E neste contexto que foi apresentado à comunidade RYR1, durante a Conferência Internacional de Famílias, realizada em julho deste ano em Pittsburgh, E.U.A., o Ryan, o novo mascote da Fundação RYR1, o qual foi calorosamente bem recebido por toda comunidade RYR1.
A fundação informou que a partir de agora, Ryan será sempre apresentado em boletins informativos, mídias sociais e site da organização, envolvendo-se em uma variedade de atividades. Ryan toca piano, pinta e participa de esportes, incluindo boliche adaptativo, passeios a cavalo, hóquei e muito mais.
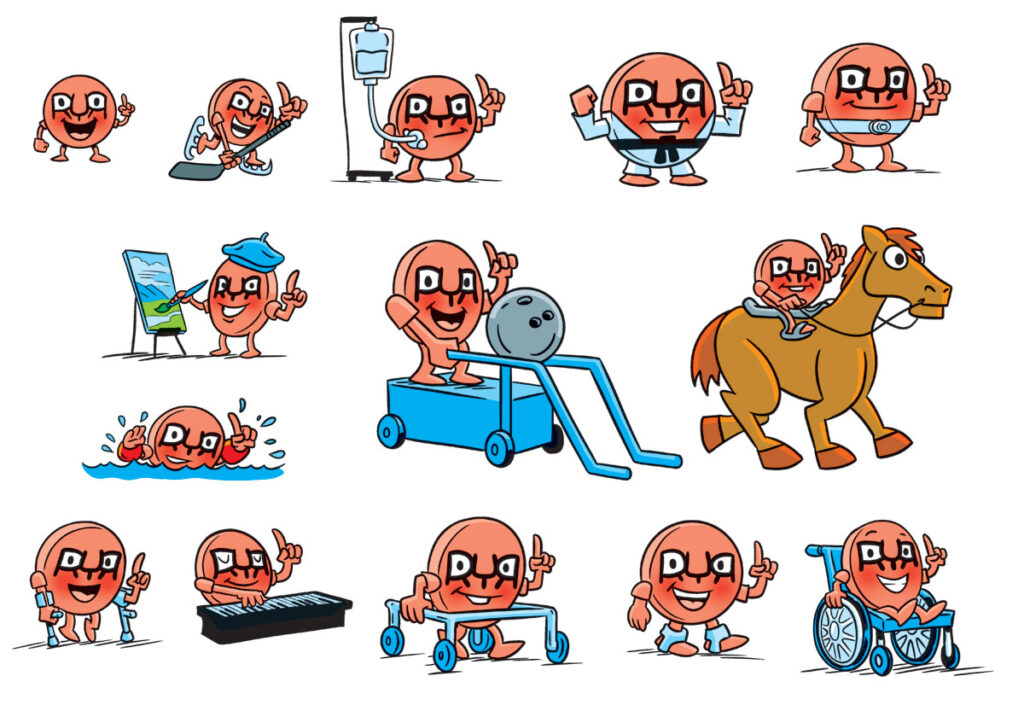
O nome "RYAN" é um aceno para o receptor de ryanodina, que é codificado pelo gene RYR1, gene responsável pelas doenças relacionadas ao RYR1. Ryan é retratado segurando um dedo para cima para representar o número "1,", enquanto as bochechas vermelhas do personagem simbolizam sensibilidade ao calor, um sintoma comum entre os membros da comunidade RYR1.
O conceito de mascote foi trazido à vida por voluntários dedicados da comunidade RYR-1-RD, incluindo integrantes do conselho administrativo e consultivo, e pelos pais Macie Soler-Sala e AJ Warren, ambos diretores criativos da Weiden+Kennedy e Goodby Silverstein & Partners, respectivamente, os quais desempenharam um papel fundamental no desenvolvimento de Ryan.
Os desenhos de Ryan foram criados por Chris Griarusso, um reconhecido artista e escritor indicado ao prêmio Harvey conhecido pela série G-Man na Image Comics e Mini Marvels para a Marvel Comics.
Ryan ganhou também uma música com um tema cativante, “Strength in Numbers”, que⇓ quer dizer fortes em número ou quantidade, ou tipo assim, a união é que faz a força, que foi escrita e interpretada por Macie Soler-Sala.
⇓ letra da música ⇓
♫ ♫ ♫ S-t-r-o-n-g (F-o-r-t-e)
Find out what it means to me (Descubra o que isso significa para mim)
Hope is in our DNA (A esperança está no nosso DNA)
We walk, we wheel, we skate, we play (Nós andamos, nós rodamos, nós patinamos, nós brincamos)
You got my back and I’ve got yours (Você está comigo e eu estou com você)
And every day we’re stronger than we were before (E cada dia somos mais fortes do que éramos antes)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
We got strength in numbers, you and me (Nós temos força em número, você e eu)
We never get tired of trying our best (Nunca nos cansamos de tentar o nosso melhor)
Until we have a cure, we just won’t rest (Até que tenhamos uma cura, não descansaremos)
So let’s raise our voices, let’s shout it out loud (Então vamos levantar nossas vozes, vamos gritar bem alto)
Every step forward is a reason to be proud (Cada passo à frente é motivo de orgulho)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
We got strength in numbers, you and me (Nós temos força em número, você e eu)
Together we’re better, together we’re stronger (Juntos somos melhores, juntos somos mais fortes)
What makes us different, makes us belong (O que nos torna diferentes, nos faz pertencer “ser aceitos”)
One, two, three, four (Um, dois, três, quatro..)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
We got strength in numbers, you and me (Nós temos força em número, você e eu)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
All I have on ain’t got nothing on me (Tudo o que tenho não tem nada contra mim)
S-t-r-o-n-g (F-o-r-t-e)
We got the power like you can’t believe (Nós temos um poder que você não pode acreditar)
S-t-r-o-n-g (F-o-r-t-e)
We got strength in numbers, you and me (Nós temos força em número, você e eu) ♫♫♫
"Estamos empolgados em receber oficialmente Ryan na comunidade RYR1", disse a Diretora Executiva, Ligação com Pacientes e Cofundadora, Lindsay Goldberg. "Muitos na comunidade podem se identificar com diferentes aspectos de Ryan, que incorpora o espírito de ser ativo, engajado e resiliente. Você verá mais do Ryan em breve!"
Desde que nasci, eu fui desafiado pelos sintomas de uma doença que me fazia ser diferente de outras crianças, ela me confrontava a todo tempo, me causava limitações físicas funcionais progressivas, e assim, causava a mim e à minha família um turbilhão de questionamentos e dúvidas sobre do que se tratava. Diante dos diagnósticos e prognósticos que recebia, dos mais piores possíveis, assim como pela falta de uma solução com um possível tratamento, eu, com toda minha inocência e intuito, resolvi que a melhor coisa que tinha a fazer era viver, e viver com intensidade, buscando a superação das minhas limitações, potencializando assim minha capacidade com o que tinha de físico, emocional, e intelectual. E sempre digo que apesar de tudo, eu nunca me vi no espelho como uma pessoa diferente, e neste contexto de vida, brinquei, namorei, me formei, casei, trabalhei muito, tive dois filhos, enfim, enquanto isso o mundo foi girando, a ciência descobrindo e explicando as coisas, até que quando tinha 44 anos, obtive meu diagnóstico sobre minha doença, ou seja, que tinha uma mutação no gene RYR1, e que me causava uma doença chamada de Miopatia Congênita Centronuclear, e que isso é que era a responsável pela condição que vivia desde que nasci. Pois bem, não mudou nada na minha vida, a não ser que descobri o endereço e nome da minha doença, o resto foi que ela era uma "Doença Rara", incurável e sem tratamento, e por fim, que pouco se conhecia sobre ela no meio médico-científico.
Sempre fui uma pessoa muito resiliente, que não só tem a força interior para enfrentar a realidade, por mais dura que seja, mas também tem a ambição intelectual e pessoal para moldar ativamente minha realidade em uma vida melhor, ou como diz minha mãe, mais "suave". Sempre fui uma pessoa da mente muito aberta ao conhecimento, ao questionamento, à observação, ao aprendizado, e essas minhas características não me fizeram ser uma pessoa acomodada. Não saber a princípio qual era doença que me acometia, e posteriormente descobri-la, e o pior que não tinha cura ou tratamento, nunca me causou sentimentos negativos, muito pelo contrário, me impulsionou a buscar viver melhor, mas também ao conhecimento.
essas minhas características não me fizeram ser uma pessoa acomodada. Não saber a princípio qual era doença que me acometia, e posteriormente descobri-la, e o pior que não tinha cura ou tratamento, nunca me causou sentimentos negativos, muito pelo contrário, me impulsionou a buscar viver melhor, mas também ao conhecimento.
A busca pelo conhecimento teórico sobre o mecanismo de funcionamento do gene, o RYR1, e como ele interferia no meu corpo foi um grande marco para meu conhecimento intelectual, assim como satisfação interna para minhas ansiedades. Contudo, o conhecer de perto, praticamente quase podendo o “tocar”, foi uma de minhas maiores experiências que já tive nesta minha peregrinação em busca de saber sobre a doença que me acomete desde que nasci. E isso se deu, depois de muito tempo, especificamente, recentemente, na última Conferência de Família e Workshop de Pesquisa do RYR1, realizada em julho de 2025 em Pittsburgh, EUA, durante uma reunião particular com o Dr. Filip Van Petegem. Na oportunidade, pude assistir um vídeo com a reconstituição em 3D “do meu RYR1”, ao mesmo tempo que o escutava fazer uma explanação específica sobre minha mutação. Dr. Van Petegem lidera um laboratório de pesquisa no Departamento de Bioquímica e Biologia Molecular na Universidade da Colúmbia Britânica (UBC), Vancouver, Canadá, no qual utiliza a cristalografia de raios X e microscopia crioeletrônica para estudar suas estruturas 3D, o que proporciona estudar a estrutura e função dos canais iônicos, com foco no músculo cardíaco e esquelético, e isso inclui o Receptor de Ryanodine (RyR). Um dos trabalhos do Dr Petegem consiste na importante abordagem em determinar as estruturas tridimensionais muito detalhadas desses canais do RYR1, permitindo a análise dos efeitos diretos das mutações na estrutura, e consequente interferência física no indivíduo afetado.
A título ilustrativo e como exemplo, gostaria de compartilhar com o leitor dessa postagem um pequeno vídeo criado pelo Dr Van Petegem, a partir da técnica acima descrita, da variante do meu RYR1 com a respectiva mutação genética. O vídeo lhe permitirá entender melhor a complexidade da estrutura molecular do gene RYR1, descrita na postagem anterior, O GIGANTE DO GENOMA QUE FAZ OS MÚSCULOS SE MOVEREM, e que neste caso se trata do meu RYR1.
O Vídeo
Para orientar um pouco melhor a apresentação deste vídeo, e assim entender o que acontece com meu gene RYR1, veja a seguir algumas da partes importantes da apresentação:
1). cada esfera que você vê representa um átomo individual;
2). o vídeo começa com uma visão 'top', ou seja, por cima do RYR1. Olhando no fundo da parte central, está a região chamada de 'poro', onde os íons de cálcio podem passar;
3). o vídeo amplia a localização da minha variante, E1175K. O aminoácido do 'tipo selvagem' é o chamado 'E', também conhecido como 'Glu' ou 'Glutamato'. Este aminoácido tem a propriedade especial de carregar carga negativa. No vídeo, este E1175 é mostrado em vermelho;
4). pode-se ver que o E1175 está apontando para outro aminoácido em uma cor diferente, em azul. Esse aminoácido, conhecido como 'R' ou 'Arg' ou 'Arginina', é especial porque carrega uma carga positiva. Assim, em pessoas sem nenhuma variante RYR1, haverá normalmente uma atração entre o 'E' carregado negativamente em vermelho, e o 'R' carregado positivamente em azul;
5). na variante demonstrada (a minha no caso), o E1175 carregado negativamente é substituído por 'K'. Este aminoácido, também conhecido como 'Lys' ou 'Lysine', também carrega carga positiva. Portanto, a variante não apenas abole a interação positivo-negativa normal, mas também coloca um aminoácido carregado positivamente (K) ao lado de outro (R), e isso desestabiliza a proteína. Por causa disso, o RYR1 se torna mais móvel e o poro pode abrir mais facilmente, ou seja, não funcionar da maneira correta. Esse defeito leva a um 'vazamento' de cálcio, que pode danificar o funcionamento normal dos músculos.
(VIDEO 1)
(VIDEO 2)
O RYR1 é um dos maiores genes do corpo humano, e é responsável para o funcionamento dos nossos músculos.
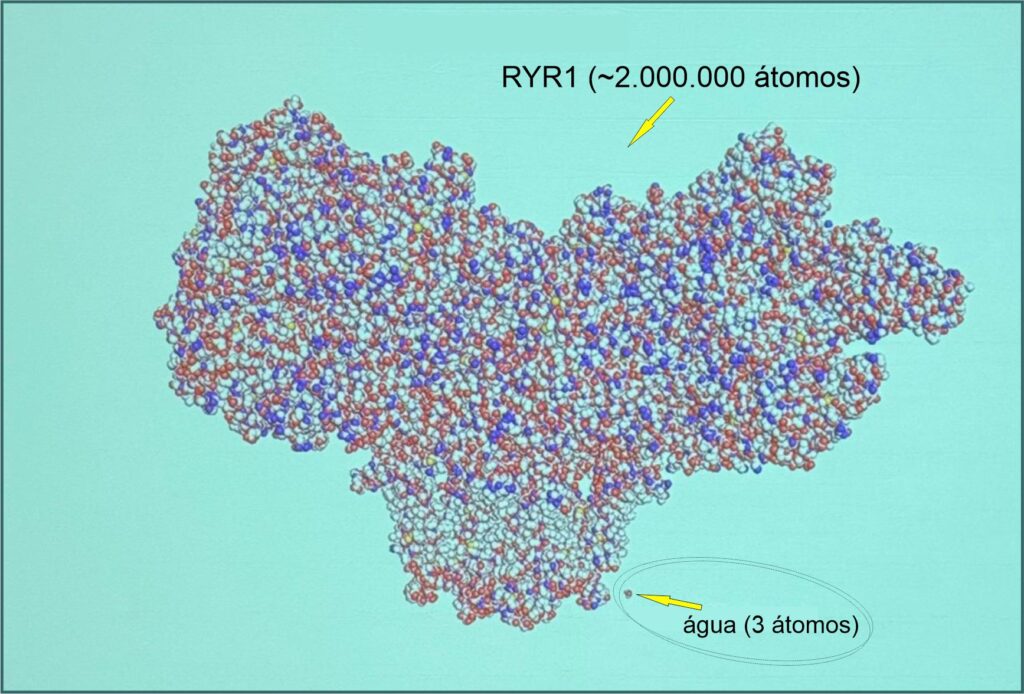
O gene RYR1 (Receptor de Rianodina 1) é um dos maiores e mais complexos genes do genoma humano. Ele contém mais de 100 exons e cerca de 15 mil pares de bases na sua sequencia de DNA, abrangendo uma grande extensão no cromossomo 19. A estimativa é que o gene RYR1 tenha cerca de 2 milhões de átomos, e esse é somente um cálculo de aproximação, pois a quantidade exata pode variar dependendo da sequência específica de nucleotídeos.
Para se ter uma idéia da complexidade do RYR1, ele é imensamente maior e mais complexo do que moléculas simples como da água, tão crucial para nossa vida, e a título de comparação, pasmem ! …o RYR1 tem 2 milhões de átomos, e a água, também conhecida como H20, tem somente 3 átomos.
O gene RYR1 que é crucial para a função do receptor de rianodina, que é um canal de cálcio localizado no Retículo 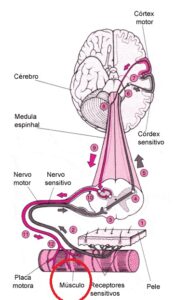 Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+
Sarcoplasmático (RS) das células musculares esqueléticas, e desempenha um papel importante no controle de liberação de íons de cálcio para dentro das células musculares, essencial no mecanismo de funcionamento do músculo, especificamente fundamental para o Acoplamento Excitação-Contração (AEC), processo de contração e relaxamento muscular. O processo funciona assim: 1). O sinal nervoso (potencial de ação) atinge o receptor DHPR na membrana da célula; 2). O DHPR, que está ligado mecanicamente ao RYR1, ele é acionado e abre o canal RYR1; 3). O RYR1 aberto libera grandes quantidades de cálcio (Ca2+) do RS para o citoplasma; 4). O cálcio dispara a contração muscular ao permitir a interação Actina-Miosina; 5). Para o relaxamento, o sinal cessa, o RYR1 se fecha, e rapidamente retorna o cálcio do citoplasma, devolvendo-o ao RS.
Em resumo, o RYR1 atua como o principal portão de saída do cálcio do reservatório intracelular. Ele é o mediador essencial que garante que o comando elétrico do nervo seja traduzido em movimento mecânico, controlando a entrada abrupta de cálcio que inicia a contração e, consequentemente, permitindo que a sua reabsorção finalize o processo para o relaxamento.
Como disse no início deste texto, devido ao tamanho e complexidade do RYR1, mutações nesse gene podem estar associadas a várias doenças musculares, incluindo:
O estudo científico do RYR1 é importante para entender essas condições ou doenças, e desenvolver tratamentos adequados em busca de minimizar seus efeitos e sintomas, pensando até na sua própria cura. Além disso, a análise genética pode ajudar em diagnósticos precoces e na prevenção de complicações associadas a essas doenças.
Durante o tempo que tenho acompanhado a movimentação de informações em torno das questões relacionadas ao RYR1, observo um grande esforço da comunidade científica em busca de entender o mecanismo de funcionamento deste complexo e grandioso gene, grande no tamanho, mas sobretudo grande em sua importância. Essas questões fazem com que o desafio ainda seja maior não só por conta dos indivíduos que têm uma doença a ele relacionada, mas também para compreensão sobre o impacto do envelhecimento celular em qualquer outro indivíduo. Observo também um grande movimento de trabalhos sendo desenvolvidos por cientistas de toda parte do mundo em busca de desenvolver uma droga para tratamentos adequados, para de repente ao menos minimizar seus efeitos e sintomas, até sua cura das doenças relacionadas ao RYR1. Particularmente, minha percepção é que as pesquisas através da técnica de reposicionamento de drogas, também chamado de redirecionamento de fármacos têm se mostrado mais promissoras no curto prazo. O “reposicionamento de drogas” se trata de uma estratégia que busca descobrir novas aplicações terapêuticas para medicamentos já existentes, que originalmente foram desenvolvidos para tratar outras doenças. Essa técnica tem a vantagem por seus reduzidos custos, assim como pela redução de tempo gasto no processo de desenvolvimento, e menores riscos no uso da eventual droga, pois já se conhece a farmacocinética e toxicidade do medicamento pesquisado, um exemplo real e prático de pesquisa em curso está se dando com o Sulfato de Salbutamol.
Contudo, a terapia genética também tem se mostrado uma abordagem promissora para o tratamento e cura de diversas doenças genéticas, oferecendo a possibilidade de corrigir ou substituir genes defeituosos. Embora este tipo de tratamento ainda esteja em desenvolvimento e não seja uma solução universal para algumas doenças, como por exemplo certas formas de miopatias ou doenças hereditárias, os resultados das pesquisas têm sido encorajadores. No entanto, a eficácia e a segurança da terapia genética podem variar dependendo da doença, do tipo de terapia utilizada, e condições do paciente. Além disso, questões éticas e de custo também são consideradas importantes. Contudo, enquanto a terapia genética oferece uma expectativa otimista, é uma área em evolução que requer mais pesquisas, altos recursos financeiros, e testes clínicos para se consolidar como a melhor solução para todas as doenças genéticas.
No caso específico das doenças relacionadas à mutação do gene RYR1, a terapia genética apresenta vários desafios, principalmente, como escrito no início deste texto, devido ao tamanho do gene, complexidade das doenças relacionadas a ele, mas também por considerar que os músculos representam aproximadamente 40% da massa total do corpo humano, e a maior parte dessa massa muscular é composta por músculos esqueléticos. Observe a seguir algumas das dificuldades:
Esses desafios tornam a pesquisa e o desenvolvimento de terapias para mutações no gene RYR1 complexos e exigem abordagens inovadoras e multidisciplinares.
Eu, enquanto portador de uma doença causada pela mutação nesse “grandioso” RYR1, tenho uma relação muito particular com esse gene, e a cada dia que passa o conheço um pouco mais, ..... confira minha próxima postagem intitulada “Eu vi o meu gene em 3D e entendi o que acontece dentro de mim”.
O SORRYR-1 mais uma vez cumpriu com seu objetivo de informar sobre as doenças relacionadas ao RYR-1 aos indivíduos afetados pela doença e seus familiares, profissionais da área da saúde, além de apoiar e participar de pesquisas científicas. Assim como já foi relatado nas redes sociais, eu fui convidado pela Dra Lizan Stinissen, do departamento de neurologia da Radboud University Medical Center, de Nijmegen, Holanda, para colaborar na elaboração de uma pesquisa internacional dirigida a pacientes com doenças relacionadas ao RYR1 visando sua participação em ensaios clínicos. O trabalho de elaboração da pesquisa foi concluído, pesquisa aplicada, e seu resultado foi apresentado pela Dra Lizan durante a Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos, tendo no final da apresentação, o orgulho de ouvir palavras de reconhecimento pela nossa participação e contribuição. O estudo se concentrou no que as pessoas com Miopatias Congênitas RYR1 e seus familiares esperam de futuros ensaios clínicos, e o que os incentivaria a participar dessas pesquisas. O objetivo deste estudo é fornecer aos pesquisadores clínicos, às empresas farmacêuticas, e à comunidade de indivíduos afetados pelas doenças relacionadas ao RYR-1, subsídios ao desenvolvimento de pesquisa em ensaios clínicos que sejam significativas e de importância terapêutica para os pacientes. O resultado da pesquisa fornecerá também aos profissionais de saúde uma visão sobre os sintomas mais importantes para os pacientes e as lacunas nas terapias disponíveis para os indivíduos com miopatia relacionada ao RYR1.
Medical Center, de Nijmegen, Holanda, para colaborar na elaboração de uma pesquisa internacional dirigida a pacientes com doenças relacionadas ao RYR1 visando sua participação em ensaios clínicos. O trabalho de elaboração da pesquisa foi concluído, pesquisa aplicada, e seu resultado foi apresentado pela Dra Lizan durante a Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos, tendo no final da apresentação, o orgulho de ouvir palavras de reconhecimento pela nossa participação e contribuição. O estudo se concentrou no que as pessoas com Miopatias Congênitas RYR1 e seus familiares esperam de futuros ensaios clínicos, e o que os incentivaria a participar dessas pesquisas. O objetivo deste estudo é fornecer aos pesquisadores clínicos, às empresas farmacêuticas, e à comunidade de indivíduos afetados pelas doenças relacionadas ao RYR-1, subsídios ao desenvolvimento de pesquisa em ensaios clínicos que sejam significativas e de importância terapêutica para os pacientes. O resultado da pesquisa fornecerá também aos profissionais de saúde uma visão sobre os sintomas mais importantes para os pacientes e as lacunas nas terapias disponíveis para os indivíduos com miopatia relacionada ao RYR1.
Veja a seguir como tudo se deu...
Pesquisa com Paciente com Doença Relacionada ao RYR1
Este estudo se concentra sobre as preferências de ensaios clínicos de indivíduos com doenças relacionadas ao RYR1 (RYR1-RD).
Antecedentes e Objetivo do Estudo
Atualmente, existem vários estudos pré-clínicos e estudos de história natural em andamento sobre o RYR1-RD, assim como já houve dois ensaios clínicos até agora. Os possíveis próximos ensaios clínicos que estão por vir indicam ter bons resultados. A comunidade de pacientes pode contribuir para esses futuros ensaios apontando suas preferências e expectativas. Espera-se que este estudo possa fornecer aos pesquisadores clínicos, empresas farmacêuticas, e à comunidade de pacientes informações de importância significativas para pesquisas terapêuticas aos pacientes. O estudo destaca os maiores sintomas e queixas, buscando identificar medidas de relevantes resultado.
Este estudo é uma continuação de uma publicação sobre uma pesquisa online com pacientes iniciada pela Fundação RYR-1, combinada com depoimentos de pacientes apresentados durante o Workshop Internacional de Pesquisa realizado em 2022. O relatório completo do estudo anterior intitulada como: “Indivíduos e Famílias Afetados por Doenças Relacionadas ao RYR1: A Perspectiva do Paciente/Cuidador”, pode ser encontrado disponível no seguinte link aqui: https://ryr1.org/medical-literature/individuals-and-families-affected-by-ryr1-related-diseases-the-patient-caregiver-perspective
Como o Estudo Foi Estruturado
A pesquisa com pacientes RYR1-RD esteve disponível online entre 10 de março e 5 de maio de 2025. Ela incluiu perguntas sobre a condição dos participantes e como ela estava sendo tratada na época, além de suas opiniões e expectativas em relação às pesquisas e ensaios clínicos. A pesquisa foi disponibilizada em 6 idiomas: inglês, espanhol, português, francês, alemão e holandês. Como apoio ao processo de tradução e revisão da pesquisa, contamos com a colaboração de perto de membros da Fundação RYR-1 e palestrantes de acordo com o seu respectivo idioma, muitos dos quais eram também pacientes.
Primeiros Resultados e Próximos Passos
Os resultados fornecem informações sobre:
Um total de 152 pessoas, representando 19 países diferentes, responderam a pesquisa. Os resultados foram apresentados na Conferência Internacional da Família promovido pela Fundação RYR-1 em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos. No momento, estamos escrevendo um artigo científico com base nos resultados da pesquisa. Planejamos submetê-lo a uma revista médico-científico, para que tanto médicos quanto pesquisadores possam se beneficiar dos resultados. Esperamos que os resultados aumentem a compreensão dos pacientes sobre sua participação futura em pesquisas clínicas, assim como apoie os pesquisadores no desenvolvimento futuros de seus projetos de pesquisas clínicas. O artigo demonstrará a importância de refletir sobre as experiências, pensamentos e expectativas dos pacientes, e destaca a realização de um trabalho de cocriação com pesquisadores acadêmicos e a comunidade de pacientes. Uma atualização seguirá assim que o artigo for finalizado.
em recente, 26 de julho de 2025 em Pittsburgh, nos Estados Unidos. No momento, estamos escrevendo um artigo científico com base nos resultados da pesquisa. Planejamos submetê-lo a uma revista médico-científico, para que tanto médicos quanto pesquisadores possam se beneficiar dos resultados. Esperamos que os resultados aumentem a compreensão dos pacientes sobre sua participação futura em pesquisas clínicas, assim como apoie os pesquisadores no desenvolvimento futuros de seus projetos de pesquisas clínicas. O artigo demonstrará a importância de refletir sobre as experiências, pensamentos e expectativas dos pacientes, e destaca a realização de um trabalho de cocriação com pesquisadores acadêmicos e a comunidade de pacientes. Uma atualização seguirá assim que o artigo for finalizado.
RYR1-RD patient survey_LizanStinissen






O entendimento das pessoas evolui à medida que a ciência avança, e novas descobertas são feitas. Nesta dinâmica, o avanço médico e científico ao longo dos últimos anos teve um impacto significativo no diagnóstico e na percepção médica sobre as doenças.
Com o desenvolvimento de novas tecnologias, métodos de pesquisa e compreensão biológica, o conhecimento genético avançou de maneira notável, especialmente após se ter conseguido fazer o o mapeamento do genoma humano. Agora, através do resultado genético é possível não só diagnosticar uma doença, mas entender seu impacto no indivíduo, e adaptar as abordagens com base no perfil genético de cada paciente.
Nestes meus mais de 60 anos, senti na pele essa grande reviravolta na ciência, no que diz respeito ao conhecimento médico e diagnóstico da minha doença. Vivi até meus 44 anos, com vários diagnósticos errados, abordagens diferentes, até que tive finalmente o diagnóstico sobre a doença que me afeta, a Miopatia Congênita Centronuclear (MCC). Essa doença é causada pela mutação no gene RYR1, não tem cura, nem tratamento. Apesar da dureza sobre as informações sobre a doença, ter o diagnóstico de certa forma me trouxe uma “espécie de alívio“. O RYR1 é responsável pelo controle do fluxo de íons de cálcio para dentro da célula muscular, que faz o músculo contrair e relaxar.
Naquela época, o resultado histológico, feito por uma biópsia muscular, era tido como sendo uma informação importante no diagnóstico, e acompanhamento clínico, porque através dele podia-se saber e diferenciar se o indivíduo era afetado por exemplo, por Miopatia Congênita Centronuclear (MCN), Miopatia Central Core (MCC), Doença Multi-Minicore (DMm), ou Desproporção Congênita do Tipo de Fibra (DCTF). Aconteceu que através de minha vivencia em meio ao pequeno mundo dos afetados por essa doença, pude ver diferentes pessoas afetadas por MCN com a evolução diferente, assim como comparados com indivíduos em pior condições físicas em relação a um outro com MCC, ou DCTF, da mesma verificando também no inverso.
O avanço das técnicas de diagnóstico genético, nos trouxe uma mudança significativa, permitindo identificar diretamente variantes no gene RYR1, possibilitando uma visão mais precisa e específica da doença. Essa abordagem é preferível porque pode confirmar a presença de alterações no gene, mesmo na ausência de características histopatológicas distintivas, permitindo uma melhor orientação do prognóstico e o acompanhamento clínico do paciente.
Passado um tempo, descobri que no diagnóstico da doença, a histologia não tem um papel tão relevante quanto anteriormente, mas sim a herança e variação genética, em que é separado as formas recessivas e dominantes. Clinicamente observa-se evolução clínica com quadros mais graves em indivíduos com herança recessiva.
Em recente conversa com Michael F. Goldberg, MD, MPH, Presidente do Conselho, Co-Presidente de Pesquisa e Co-Fundador da @theryr1foundation, ele me disse: “Casos recessivos de RYR-1 tendem a ser mais graves quando associados a variantes do gene RYR1 resultando em expressão reduzida da proteína receptora RYR-1. As formas dominantes tendem a ser menos graves porque essas variantes geralmente não resultam em quantidades reduzidas da proteína receptora RYR-1. Os diagnósticos histopatológicos derivados da biópsia muscular (por exemplo, Centronuclear, Central Core, ou DCTF, etc.) não parecem ser muito informativos e não são específicos para fenótipos específicos mesmo para RYR1. Portanto, houve um afastamento do diagnóstico histopatológico e uma ênfase maior no diagnóstico genético .”
Em resumo, os diagnósticos histopatológicos obtidos a partir de biópsias musculares, como Centronuclear, Central Core, Desproporção Congênita do Tipo de Fibra, ou Multi Mini Core, não são muito informativos, e não são específicos para certos fenótipos. Isso levou a uma mudança para um maior enfoque no diagnóstico genético, que é mais preciso e informativo. Esse avanço no entendimento científico, permitiu a identificação e conhecimento das variantes genéticas, e sua relação com a gravidade das doenças associadas ao gene RYR1, crucial para um diagnóstico eficaz, e para o desenvolvimento de abordagens adequadas ao paciente.
A Miopatia Congênita Centronuclear é uma condição neuromuscular rara, com características de progressividade, e que pode levar a diversas complicações, incluindo problemas ortopédicos, como já relatado em postagem anterior (clique aqui), mas também podem ocorrer complicações respiratórios. As questões respiratórias podem surgir devido à fraqueza muscular que afeta os músculos respiratórios, resultando em dificuldades na ventilação e na troca gasosa. Para melhor entender o contexto desta questão das complicações respiratórias em indivíduos com miopatia relacionada ao RYR-1, o texto a seguir será dividido em tópicos conceituais e explanatórios.
OXIGÊNIO E O SISTEMA RESPIRATÓRIO
O oxigênio é de vital importância para o corpo humano, especificamente a cada célula, órgão e sistema do organismo. No caso dos músculos, o oxigênio desempenha um papel fundamental, como segue:
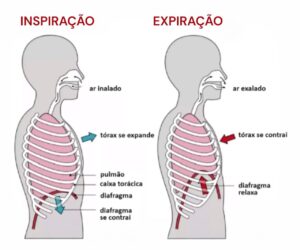 O sistema respiratório, através do processo de respiração, é o responsável por alimentar todo nosso oragnismo de oxigênio, elemento tão precioso e responsável por nos manter vivos. A respiração é um processo involuntário e automático que ocorre em duas etapas: Inspiração e Expiração.
O sistema respiratório, através do processo de respiração, é o responsável por alimentar todo nosso oragnismo de oxigênio, elemento tão precioso e responsável por nos manter vivos. A respiração é um processo involuntário e automático que ocorre em duas etapas: Inspiração e Expiração.
Trazendo toda essa questão da respiração para o contexto de um portador de uma miopatia, deve-se destacar que o diafragma é o principal músculo respiratório. O processo respiratório funciona assim:
RESPIRAÇÃO EM UM INDIVÍDUO COM MIOPATIA
Um indivíduo com miopatia tende ter fraqueza no diafragma e nos músculos abdominais, e essa situação pode dificultar a desobstrução das vias aéreas, pois se torna difícil inspirar profundamente e expirar de forma forte e completa. Se por exemplo um indivíduo com miopatia pega um resfriado, com seus músculos respiratórios enfraquecidos, ele pode desenvolver uma pneumonia. Esse processo inflamatório nos pulmões pode enfraquecer ainda mais os músculos e causar mais problemas na desobstrução das vias aéreas. A parede torácica e o abdômen formam a caixa torácica. Se a parede torácica estiver fraca, durante a inspiração, o tórax pode se movimentar para dentro em vez de se mover para fora. Isso dificulta ainda mais a respiração profunda. À medida que a parede torácica se enfraquece e se movimenta menos, ela também fica rígida. A rigidez da parede torácica torna ainda mais difícil a respiração profunda.
A fraqueza da caixa torácica também pode causar instabilidade da coluna. Isso pode causar uma curvatura na coluna, o que resulta em cifose ou "corcunda" nas costas. Essa instabilidade também pode causar a curvatura lateral da coluna, o que resulta em escoliose. Essas curvaturas podem limitar o movimento da parede torácica durante a respiração, o que dificulta a respiração profunda e resulta em menor volume dos pulmões. Juntos, esses problemas podem levar a uma situação em que os músculos respiratórios não funcionam bem o suficiente para trazer oxigênio para dentro do corpo e eliminar o dióxido de carbono. Isso se chama insuficiência respiratória.
COMPLICAÇÕES RESPIRATÓRIAS EM UM INDIVÍDUO COM MIOPATIA
Assim como ocorre com outros sintomas nos indivíduos afetados por uma das doenças relacionadas ao RYR-1, a gravidade das complicações respiratórias também varia de indivíduo para indivíduo. Assim, algumas pessoas com doenças relacionadas ao RYR-1 podem não ter problemas. Alguns podem ter problemas leves, mas precisam de auxílio para respirar durante o sono ou quando estão doentes. Em casos graves, alguém com doença relacionada ao RYR-1 pode precisar de ventilação mecânica para respirar. Segue algumas das complicações respiratórias que podem ocorrer incluem:
É importante que os indivíduos com Miopatia Congênita Centronuclear sejam monitorados de perto quanto à função respiratória e recebam cuidados multidisciplinares para gerenciar eventuais complicações. O diagnóstico preciso de complicações respiratórias envolve uma variedade de exames que permitem aos médicos avaliar a função pulmonar, iniciando por um exame clínico, raio-x, tomografia computadorizada, teste de função pulmonar (TFP), polissonografia, exames laboratoriais, dentre outros. Uma vez identificado uma complicação respiratória, o tratamento pode incluir fisioterapia respiratória, suporte ventilatório e acompanhamento regular com especialistas em pulmão. E caso o indivíduo, afetado por Miopatia Centronuclear pela mutação no RYR-1, necessite de hospitalização, é importante se ter preventivamente um plano de suporte respiratório e orientação clínica. Na necessidade de uma cirurgia, os médicos deverão adotar precauções para suporte respiratório antes e depois da cirurgia, além de serem alertados sobre a sussetibilidade à Hpertermia Maligna.
ABORDAGENS RESPIRATÓRIAS PREVENTIVA E TERAPEUTICA EM UM INDIVÍDUO COM MIOPATIA¹
Assim como relatado em postagem anterior Abordagem Respiratória na Miopatia Centronuclear (clique aqui) a fisioterapia respiratória é responsável pela minimização dos comprometimentos respiratórios nas miopatias centronucleares, pois interfere na progressão da perda muscular, minimiza as complicações da perda de capacidade pulmonar, mantem a funcionalidade respiratória como fala, deglutição e tosse. Fazem parte das estratégias utilizadas no tratamento:
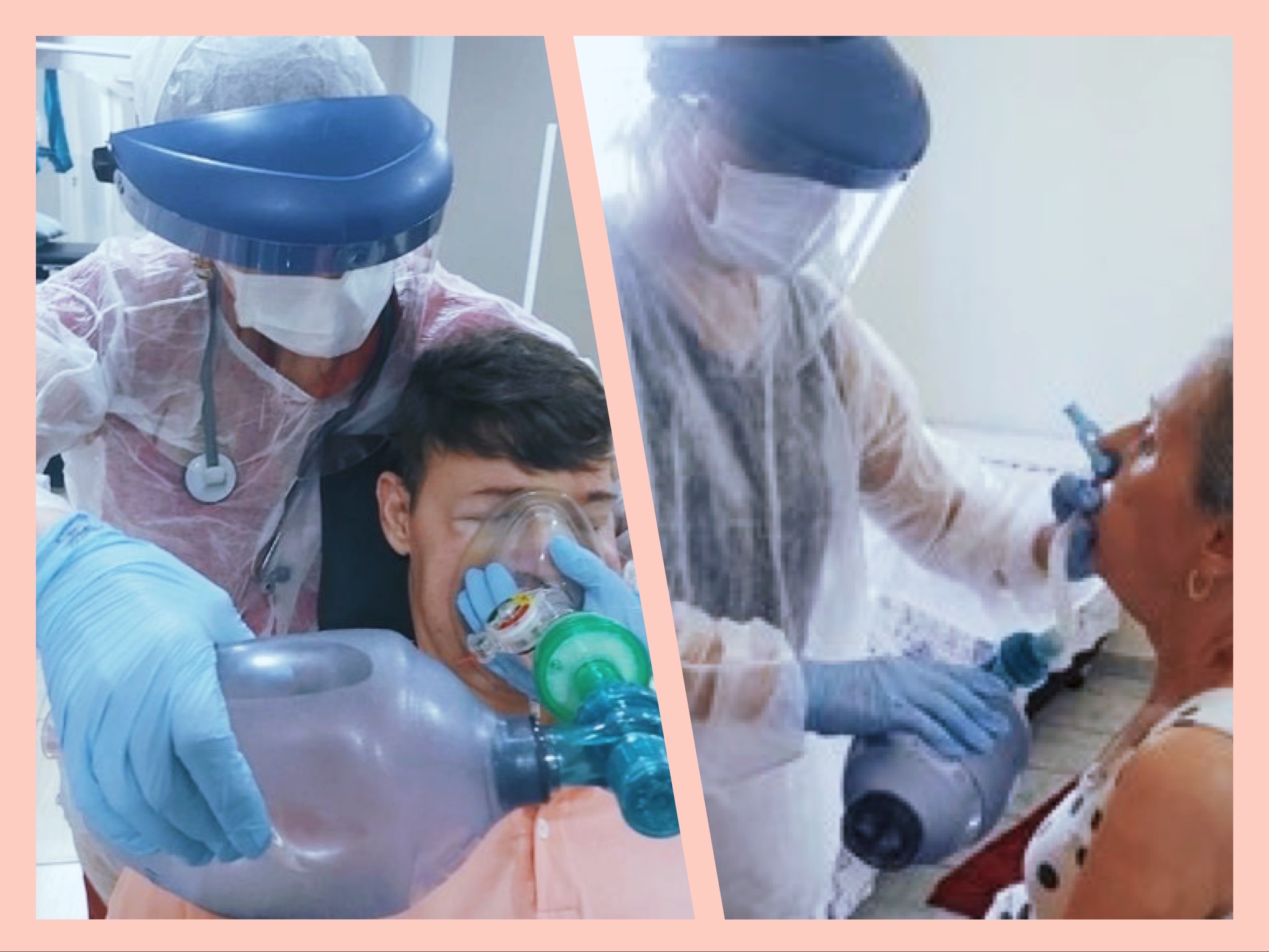
Fig 1 -Exercício de empilhamento de ar \ Fig 2 - Exercícios respiratórios para manutenção da capacidade pulmonar e treinamento da musculatura bulbar (ambos utilizando a bolsa de insuflação (ambu)
O cuidado respiratório deve ser uma prioridade nos portadores de Miopatia Centronucelar, e o profissional deve ser capaz de identificar a alteração funcional e correlacionar com a melhor terapia a ser aplicada, sendo esta a melhor estratégia para evitar maiores complicações para o paciente.
¹ texto cedido por colaboração da fisioterapeuta Alessandra Dorça do Instituto Alessandra Dorça @institutoaledorca
Hans Christian, um escritor dinamarquês conhecido mundialmente por suas histórias infantis, certa vez disse, "Onde as palavras falham, a música fala", e eu completo dizendo que a música é um meio de transporte capaz de dirigir nossas emoções e sensações a todos os sentimentos. Contudo, através da música pude também experimentar outros benefícios além do que normalmente esperado, e que vou contar a seguir qual foi…
Quando Luciano (meu filho) se foi aos 23 anos de idade, eu busquei de alguma forma me manter próximo dele alimentando as lembranças sobre ele. Luciano, parece ter vivido pouco tempo de vida, sim, mas ele fez muito… ele foi triatleta, músico multi-instrumentista, dentre outros feitos. Com esta vontade que tive de buscar estar perto dele, pensei, no esporte não vou conseguir viver o que ele viveu, daí decidi ir de encontro com ele pelo caminho da música e então tocar violino, seu principal instrumento musical desde os 6 anos de idade. Música sempre foi algo que fez parte do dia a dia de nossa família, e cremos que Deus além de usar a música para falar conosco, ele nos concedeu o dom da musicalidade, daí não vi maiores dificuldades e resolvi embarcar nessa jornada. O Professor Ricardo Seoud embarcou comigo nesta grande empreitada para me ajudar a conquistar o objetivo de aprender a tocar violino, contudo eu sabia que antes iria precisar romper com minhas barreiras de dificuldade física para conseguir segurar o instrumento. Sendo assim, além do meu professor Seoud, contei com o trabalho conjunto de Lorena, minha fisioterapeuta, a qual me acompanhou fazendo as devidas orientações de técnicas de exercícios de consciência corporal, educação postural, alongamentos, e respiração.
Existe um fato interessante que liga minha relação como portador de deficiência física ao de tocar um instrumento musical. Quando era criança minha mãe me colocou na aula de violão, e mais tarde me deu uma flauta, não entendia o porque, mas e hoje pude concluir que mesmo com sua simplicidade sobre o entendimento da doença que me acometia, o que ela realmente buscava era promover a minha educação física, e com a flauta o meu condicionamento respiratório. Não existia conhecimento técnico que tocar um instrumento musical poderia funcionar como uma forma de fisioterapia. Naquela época, as minhas limitações físicas ainda não estavam tão severas como atualmente, hoje o simples fato de conseguir segurar o violino é o meu grande desafio. Lembro que nas primeiras aulas, Sissi, minha esposa, precisava colocar duas a três almofadas sob meu braço para servir de apoio de sustentação para conseguir segurar o instrumento, mas que no decorrer dos treinamentos eu superei e consegui manter o instrumento na posição correta, ou melhor, meio correta, e penso que foi Luciano quem estaria me ajudando fisicamente, não com o triathlon, mas com o simples ato de conseguir fisicamente segurar o violino.
Sabe-se através do conhecimento fisiológico, que o ato de tocar um instrumento musical envolve uma série de movimentos físicos que podem ajudar a melhorar a coordenação motora, a força muscular e a flexibilidade, além de estimular áreas do cérebro relacionadas à motricidade e à cognição. No mais, a música pode ter um efeito terapêutico, promovendo relaxamento, reduzindo o estresse e melhorando o bem-estar emocional.
É um entendimento, que toda forma de atividade física é crucial e benéfica para o condicionamento físico dos portadores de miopatia, inclusive como sendo a única e comprovada forma de tratamento. E no caso, pela minha experiência pessoal pude concluir que o ato de tocar um instrumento musical, é um exercício físico que atuou verdadeiramente como um complemento à minha rotina diária de fisioterapia proporcionando os seguintes benefícios:
Por fim, gostaria de ilustrar e celebrar esta postagem com uma música muito significativa para mim (senti A música escolhida, Londonderry Air, é uma ária publicada em 1855, e que se tornou como um hino irlandes, sendo considerada uma das canções mais tocadas no mundo, e que teve muitas versões de letras, mas a mais conhecida e autêntica foi Danny Boy, composta por um inglês em 1910. Sua melodia emotiva e letra tocante, fala sobre a despedida, e espera de um amor eterno, com uma mensagem de esperança e a promessa de que o amor por alguém persistirá além da vida.
...ao abrir o video no yoytube, click no canto direito inferior no icone ⌈ ⌉ para abrir a imagem em toda a tela
A grande maioria das pessoas a partir dos 50 anos se deparam com complicações ortopédicas, seja nas articulações, coluna, quadril, dentre outros, e isso geralmente se deve pelo uso inadequado do corpo gerando os desgastes naturais que acontecem no decorrer da vida, ou acontece também pela falta de exercícios físicos.
No caso do indivíduo portador de miopatia centronuclear, doença essa causada pela mutação no gene RYR-1, responsável pelo funcionamento dos músculos, as implicações ortopédicas em relação a uma pessoa sem a doença, é potencializada, e pode levar a uma série de complicações já desde o nascimento. Como já explicado em postagens anteriores, cada pessoa com doença relacionada ao RYR-1 é única, assim como a evolução da doença, as complicações também podem afetar diferentemente cada indivíduo.
Nesta postagem, vou discorrer sobre alguns dos principais problemas ortopédicos com os quais pessoalmente convivo, e que podem ocorrer com os indivíduos afetados por uma doença relacionada ao RYR-1, que são:
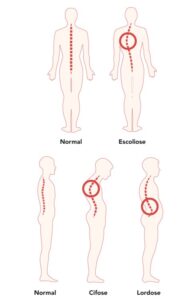
 nas articulações e tendões. No nosso caso, portadores de miopatia, a situação é logicamente agravada, porque como nossos músculos não são utilizados adequadamente, o enfraquecimento muscular é aumentado, resultando em instabilidade nas articulações, e isso pode aumentar o risco de lesões, como distensões ou rupturas de tendões. Além disso, a imobilidade por fraqueza muscular pode levar à rigidez articular e à perda de amplitude de movimento, o que pode causar dor e desconforto. Essa falta de mobilidade muscular também pode contribuir para o desenvolvimento de condições como a artrite, pois as articulações não recebem o movimento necessário para manter a saúde e a lubrificação adequada.
nas articulações e tendões. No nosso caso, portadores de miopatia, a situação é logicamente agravada, porque como nossos músculos não são utilizados adequadamente, o enfraquecimento muscular é aumentado, resultando em instabilidade nas articulações, e isso pode aumentar o risco de lesões, como distensões ou rupturas de tendões. Além disso, a imobilidade por fraqueza muscular pode levar à rigidez articular e à perda de amplitude de movimento, o que pode causar dor e desconforto. Essa falta de mobilidade muscular também pode contribuir para o desenvolvimento de condições como a artrite, pois as articulações não recebem o movimento necessário para manter a saúde e a lubrificação adequada.
Para concluir, eu diria que as complicações ortopédicas acima descritas podem ser inevitáveis aos portadores de miopatias relacionadas ao RYR1, sejam elas por origem congênita, ou em decorrência das adaptações que o indivíduo precisa fazer para conseguir movimentar durante a vida, ou pelas que acontecem com o desgaste natural do envelhecimento. Como portador de Miopatia Congênita Centronuclear, eu convivo com todas as complicações descritas, e de todas as origens de causas. Contudo, vejo que existem formas de se prevenir, postergar o seu surgimento ou agravamento, e até de se mitigar os sintomas das complicações ortopédicas. A educação postural, e o desenvolvimento pessoal de consciência corporal, devem fazer parte do nosso dia a dia. No caso de as complicações já terem se instaladas, o manejo desses problemas geralmente envolve uma combinação de fisioterapia, alongamentos, terapia ocupacional e, em alguns casos as intervenções cirúrgicas para corrigir deformidades ou melhorar a função do corpo. Por fim, é fundamental que os portadores de miopatia centronuclear sejam acompanhados por uma equipe multidisciplinar para abordar essas questões de forma abrangente, preventiva e terapêutica.

