ESPERANÇA AOS PORTADORES DE DOENÇAS RELACIONADAS AO RYR1 VISTA POR UM OUTRO LADO DO PRISMA
Na postagem anterior tratei sobre as boas notícias que tive durante o workshop científico sobre as doenças relacionadas ao RYR1 em Pittsburgh (EUA), em julho de 2022. Me referi às pesquisas sobre uma droga capaz de aliviar, tratar, e até curar as doenças relacionadas ao RYR1, mencionei inclusive sobre valores de investimentos em pesquisas e tendências de mercado para os próximos anos. Contudo pode ter ficado uma pergunta no ar, sobre o porquê do grande interesse em pesquisas tão dispendiosas sobre uma doença que atinge um pequeno número de pessoas. …então vamos buscar o raciocínio lógico…
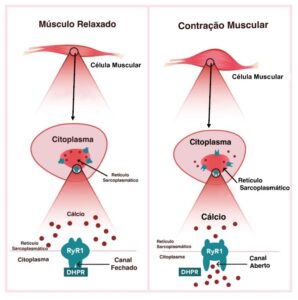
Músculo é o tecido responsável pelo movimento do corpo humano. O mecanismo de funcionamento do músculo, ou seja, como ele contrai e relaxa, é chamado de acoplamento excitação-contração (EC), e tudo acontece na célula da fibra muscular, onde o citoplasma está localizado. Lá dentro, o Retículo Sarcoplasmático (RS) atua como reservatório para íons de cálcio, essenciais para desenvolver a força muscular. O receptor de Ryanodina Tipo 1 (RYR1) funciona como canal liberador dos íons de cálcio, o qual quando liberado no citoplasma, faz com que o músculo se contraia. Na prática o mecanismo de funcionamento é simples, quanto mais íons de cálcio liberado no citoplasma, mais força muscular o indivíduo desenvolve.
O cientista Andrew R. Marks, MD, professor de fisiologia e biofísica celular na Universidade de Columbia (EUA), explica em seus estudos que a perda da função muscular é associada à disfunção dos canais de liberação do receptor de rianodina (RYR1) do músculo, e que pode ser causado por uma falha decorrente a uma mutação genética, ou devido à sobrecarga oxidativa celular relacionada à idade. Essa falha desestabiliza o estado fechado do canal, resultando em um defeito pelo vazamento de cálcio intracelular, acarretando em função muscular reduzida, o que explica não somente as doenças relacionadas ao RYR1, mas também explica no caso da oxidação celular, a razão pela qual os exercícios físicos se tornam mais difíceis com a idade.
A boa notícia é que existe uma droga em fase de testes que pode barrar esse vazamento. Na pesquisa foram estudadas as células musculares de ratos novos e velhos. Um rato de seis meses cujos receptores de rianodina vazavam cálcio mostraram os mesmos problemas de fraqueza muscular do que um rato mais velho. Este, por sua vez, apresentou melhoras depois de ser tratado com a droga em teste. O estudo sugere aos cientistas procurarem por novos caminhos no tratamento do envelhecimento. “As pesquisas que temos visto se focam em produzir mais músculos”, diz Andrew Marks. “E a diferença é que nós focamos não no músculo, mas no seu mecanismo de funcionamento, pois o aumento de músculos não ajuda se eles não funcionarem.”
Portanto, entendo que quando a comunidade científica desenvolve pesquisas em busca de uma droga capaz de tratar e até curar as doenças musculares relacionadas ao RYR1, se busca também beneficiar todos os indivíduos, porque a fraqueza muscular decorrente do envelhecimento é comum a todos. Assim, podemos pensar que o interesse mercadológico para a descoberta de uma droga capaz de atuar no defeito ou mal funcionamento da célula muscular, pode ser maior por ir de encontro com a necessidade de todos os indivíduos, e não somente aos portadores de doenças relacionadas ao RYR1. Por fim eu faço a seguinte analogia, nós portadores de uma doença relacionada ao RYR1 temos um defeito de fábrica, que é a mutação genética, e os indivíduos saudáveis terão um defeito por conta do tempo ou idade, que é a oxidação celular.
O WORKSHOP COM PESQUISADORES DE 8 PAISES DIFERENTES QUE ACONTECEU DURANTE A CONFERÊNCIA INTERNACIONAL SOBRE DOENÇAS REALCIONADAS AO RYR-1 REALIZADA EM JULHO DE 2022 EM PITTSBURGH, EUA, TROUXE GRANDES NOVIDADES SOBRE PESQUISAS EM BUSCA DE TRATAMENTO E CURA DA DOENÇA
A descoberta de qualquer doença, nunca é algo esperado, é bem desagradavel, e no caso de uma doença grave será sempre uma novidade traumática. Acontece que na grande maioria das doenças existe a opção ao afetado de um tratamento que funciona como um alento e até motivação para o novo momento que será vivido até que se alcance sua cura. Contudo, existem aquelas doenças com prognóstico evolutivo, que não contam com tratamento, muito menos cura, e que fazem com que só reste ao afetado ter que encarar a realidade do diagnóstico, e desenvolver por si só meios para conviver com suas dificuldades, sejam as físicas e as emocionais.
Recentemente participei da Conferência Internacional sobre as Doenças Relacionadas ao RYR-1, que contou com a presença de mais de 200 pessoas entre afetados e familiares, evento este que foi precedido por um workshop que reuniu 45 pesquisadores presenciais e 10 virtuais de 8 países diferentes, os quais compartilharam seus trabalhos em busca de tratamento ou cura da doença. O evento além de promover a sociabilização entre os participantes, teve como objetivo, através dos palestrantes, informar questões conceituais sobre a doença, até às novidades sobre as pesquisas para tratamentos da doença.
Como as doenças relacionadas ao RYR1 ainda não contam com tratamento, muito menos cura, foi natural observar a grande ansiedade entre os presentes em saber sobre as novidades do andamento das pesquisas em busca de uma droga capaz de aliviar sintomas e tratar as complicações causadas pela doença.
Providencialmente os cientistas palestrantes foram portadores de boas notícias que vieram de encontro com o anseio dos presentes, e informaram que estão trabalhando ativamente por meio de várias abordagens de pesquisas em busca por uma droga capaz de aliviar os sintomas da doença, contudo este é um processo bem complexo, e normalmente leva muitos anos e altos custos de investimentos.
Falando em alto custo, algo que parecia tão distante, mas que já faz parte de nossa realidade, foi pessoalmente abordado por Jacques Tremblay, PhD, cientista responsável da University of Laval, Québec, Canadá, pelas pesquisas da terapia de reposição gênica através da correção da mutação pontual no gene RYR1 usando a tecnologia de edição CRISPR/Prime editing technology. Com a “edição de genes”, o objetivo é corrigir ou “editar” apenas uma pequena parte do gene “defeituoso”, oferecendo assim o potencial de cura para doença, não apenas um tratamento.
pessoalmente abordado por Jacques Tremblay, PhD, cientista responsável da University of Laval, Québec, Canadá, pelas pesquisas da terapia de reposição gênica através da correção da mutação pontual no gene RYR1 usando a tecnologia de edição CRISPR/Prime editing technology. Com a “edição de genes”, o objetivo é corrigir ou “editar” apenas uma pequena parte do gene “defeituoso”, oferecendo assim o potencial de cura para doença, não apenas um tratamento.
Várias pesquisas de medicamentos estão em desenvolvimento como terapias potenciais para as doenças relacionadas ao RYR1 (DR-RYR1). Para mim a grande novidade foi saber que alguns desses medicamentos já estão aprovados para outras doenças, e os pesquisadores estão testando o “reposicionamento dessas drogas” para as doenças relacionadas ao RYR1 (DR-RYR1), como por exemplo, MitoQ, Dantroleno, Ácidos Graxos Poli-insaturados, Salbutamol, Piridostigmina, NAC (N-Acetil L-Cisteína), dentre outros.  Algumas destas drogas têm como alvo o próprio receptor RYR1, outras visam características e sintomas específicos das DR-RYR1. O reposicionamento de fármacos (medicamentos) consiste na técnica de pesquisa e identificação de novas indicações terapêuticas além dos já conhecidos. O seu desenvolvimento clínico requer menos tempo e menos etapas, visto que o perfil de segurança do medicamento já é conhecido, demonstrado e aprovado, assim como por ter um investimento mais baixo. A pesquisa de novas indicações terapêuticas pressupõe abordagens experimentais e computacionais que busquem identificar e gerar novas hipóteses de interações fármaco-alvo desconhecidas até então. Exemplos conhecidos de reposicionamento de fármacos é o Viagra, que foi inicialmente desenvolvido para tratar hipertensão e angina, mas que durante as etapas de estudo clínico acabou sendo direcionado para tratar disfunção sexual masculina, e outro famoso foi o caso da Zidovudina (AZT), primeiro fármaco aprovado para tratamento da AIDS, e cuja indicação original era o câncer.
Algumas destas drogas têm como alvo o próprio receptor RYR1, outras visam características e sintomas específicos das DR-RYR1. O reposicionamento de fármacos (medicamentos) consiste na técnica de pesquisa e identificação de novas indicações terapêuticas além dos já conhecidos. O seu desenvolvimento clínico requer menos tempo e menos etapas, visto que o perfil de segurança do medicamento já é conhecido, demonstrado e aprovado, assim como por ter um investimento mais baixo. A pesquisa de novas indicações terapêuticas pressupõe abordagens experimentais e computacionais que busquem identificar e gerar novas hipóteses de interações fármaco-alvo desconhecidas até então. Exemplos conhecidos de reposicionamento de fármacos é o Viagra, que foi inicialmente desenvolvido para tratar hipertensão e angina, mas que durante as etapas de estudo clínico acabou sendo direcionado para tratar disfunção sexual masculina, e outro famoso foi o caso da Zidovudina (AZT), primeiro fármaco aprovado para tratamento da AIDS, e cuja indicação original era o câncer.
Outra grande novidade foi saber sobre o RYCALS, que é uma nova classe de medicamentos que atua diretamente no receptor RYR1, estabilizando e melhorando a ligação de RYR1 e calstabina, restaurando assim a vedação para evitar vazamento de cálcio, situação essencial para o bom funcionamento da célula muscular. Pesquisadores mostraram que o tratamento com RYCALS restaura a função muscular em camundongos in vivo e no tecido muscular humano ex vivo. Especificamente, um estudo recente mostrou que RYCALS melhorou a ligação da calstabina a o receptor RYR1 em células musculares retiradas de biópsias de pacientes com doenças relacionadas ao RYR1. No final do ano de 2021, a ARMGO Pharma, desenvolvedora do RYCALS, recebeu um investimento de US$ 35 milhões de uma empresa europeia de capital de risco. Um teste clínico humano de Rycals foi concluído . O Dr. Payam Mohassel, membro da Fundação RYR-1 e investigador principal do estudo RYCALS, fez uma apresentação muito antecipada na Conferência da Família que resumiu as principais descobertas deste importante estudo. Especificamente, o estudo revelou que o RYCALS foI bem tolerado pelos pacientes sem efeitos colaterais. Embora o estudo tenha sido pequeno, demonstrou algumas tendências promissoras em testes que mediram os sintomas de fraqueza e fadiga dos pacientes. Diante dessas descobertas, o Dr. Mohassel e a equipe de desenvolvimento clínico do RYCALS acreditam que o desenvolvimento clínico adicional da droga é garantida.
Por fim, eu fico muito animado por saber que, corroborando com o relato acima descrito, de acordo com informações obtidas da Data Bridge Market Research , empresa de análise de mercado, espera-se que o mercado de pesquisas farmacêutico de doenças relacionadas ao receptor de rianodina tipo 1 (RYR1) cresça 6,40% a uma taxa anual composta (CAGR) no período de previsão de 2021-2028.
A FUNDAÇÃO RYR-1 REALIZOU EM JULHO DE 2022, EM PITTSBUGH, EUA, O PRIMEIRO WORKSHOP INTERNACIONAL DE PESQUISAS SOBRE DOENÇAS RELACIONADAS AO RYR-1
A Fundação RYR-1 realizou nos dias 21 e 22 de julho último, em Pittsburgh, nos Estados Unidos, o primeiro Workshop Internacional de Pesquisas sobre Doenças Relacionadas ao RYR-1, para discutir desde os mecanismos ao tratamento da doença. O evento reuniu 45 pesquisadores presenciais e 10 virtuais de 11 países diferentes, os quais se encontram desenvolvendo trabalhos em busca de tratamento ou cura da doença, e que na oportunidade puderam compartilhar entre eles suas respectivas pesquisas.
A organização do encontro convidou também 10 indivíduos afetados pelas doenças relacionadas ao RYR1, estando eu entre eles, para relatar nossas diferentes histórias de vida em relação à doença. O objetivo foi compartilhar nossa vivência, conhecimentos e interagir com os presentes, visando assim colaborar no desenvolvimento de novas estratégias para encontrar terapias, assim como também sensibilizar os pesquisadores sobre nosso anseio urgente por alguma forma de resultado prático de suas pesquisas para amenizar sintomas e até curar a doença.
No meu testemunho de vida diante da doença, em que relatei acontecimentos desde meu nascimento até os dias de hoje, teve três pontos que percebi chamar a atenção dos presentes. Um dos pontos foi sobre a exaustão sofrida em função do longo período de 44 anos que se levou para o diagnóstico. Essa mesma demora de tempo para diagnostico não seria uma realidade nos dias de hoje, mas ainda deve ser considerada como alta, pois sabe-se que atualmente uma “doença rara” leva em média de 7 a 10 anos para ser diagnosticada. No meu caso, a Miopatia Congênita Centronuclear não é uma das doenças mais comuns dentro do espectro das relacionadas a mutação do RYR-1, dificultando assim o seu diagnóstico. Outro ponto importante destacado foi dizer que mesmo recebendo o diagnóstico da doença, a falta de informação dado ao seu pouco conhecimento médico-científico, me causou o que disse ter sido uma espécie de cura emocional, gerando um grande estímulo positivo em busca de respostas ao desconhecido da doença. Entendo que a falta de diagnóstico e informações sobre uma doença, muitas vezes é pior do que seus próprios sintomas, e que o emocional tem um grande poder sobre o físico. E por fim, disse sobre o grande  momento vivido depois de 10 anos do diagnóstico, em que conheci a Fundação RYR-1, momento aquele em que ainda buscava informação sobre a doença. Essa organização tem o objetivo promover a troca de experiências entre os participantes, além de apoiar cientistas que trabalham no desenvolvimento de medicamentos e tratamento, a qual mostrou a mim que a Miopatia Congênita Centronuclear não era um campo tão desconhecido como pensava, e me fez acreditar em algo que eu nunca tinha pensado antes, que era a existência próxima de um medicamento para tratar ou aliviar os efeitos das Doenças relacionadas ao RYR1.
momento vivido depois de 10 anos do diagnóstico, em que conheci a Fundação RYR-1, momento aquele em que ainda buscava informação sobre a doença. Essa organização tem o objetivo promover a troca de experiências entre os participantes, além de apoiar cientistas que trabalham no desenvolvimento de medicamentos e tratamento, a qual mostrou a mim que a Miopatia Congênita Centronuclear não era um campo tão desconhecido como pensava, e me fez acreditar em algo que eu nunca tinha pensado antes, que era a existência próxima de um medicamento para tratar ou aliviar os efeitos das Doenças relacionadas ao RYR1.
Encontros como as conferências de famílias, e esse primeiro workshop de pesquisadores, me fazem entender que apesar de raro ou em meio a poucos, não me encontro sozinho, e assim além de aprender com os outros sobre como conviver com a doença, alimenta a expectativa em saber que existem médicos e cientistas em adiantado estágio de pesquisa por uma droga para tratamento e até a sua cura.
A COMUNIDADE CIENTÍFICA AFIRMA QUE ATIVIDADE FÍSICA É IMPORTANTE PARA PORTADORES DE MIOPATIA COMO FORMA TERAPÉUTICA
Sempre que estamos diante do diagnóstico de uma doença, nossa primeira reação é se informar sobre a existência de tratamento e cura. Trazendo esta situação para realidade das Doenças Relacionadas ao RYR1, não teremos resposta para ambas as questões. Contudo, sabe-se que atualmente os cientistas estão trabalhando ativamente em abordagens potenciais de tratamento e até cura, seja de uma droga a até terapia genética.
A comunidade científica afirma que atividade física é importante para os portadores de miopatia para estimular a força muscular, proporcionar o equilíbrio, promover alongamento, manter em movimento, e buscar sua independência.
Em março de 2012 foi publicado no Journal of Child Neurology a “Declaração de Consenso sobre Padrão de Cuidados para Miopatias Congênitas”, em que se recomenda sob certas circunstâncias e cuidados que se faça exercícios aeróbicos regulares, se possível pelo menos duas ou três vezes por semana.
Sabe-se também que a falta de atividade física para qualquer pessoa causa a perda de força e massa muscular, perda óssea, ganho de peso, compromete o sistema cardio-respiratório, dentre outros, agora imagina esses efeitos danosos para um portador de miopatia.
Por minha experiência de vida como portador de Miopatia Congênita Centronuclear, diria que atividade física não é somente uma forma de promover a manutenção física, mas de tratar a miopatia, pois além de proporcionar o condicionamento e fortalecimento físico, promove a prevenção de eventuais complicações causadas pela doença, podendo inclusive conter seu agravamento.
Como conclusão prática, lembro que o comportamento das pessoas durante o confinamento motivado pela pandemia de COVID-19, fez com que estas de um modo geral se isolassem como forma de distanciamento social, e por conta disso, reclusas em suas casas, reduziram consideravelmente a atividade física, aumentando o comportamento sedentário, e tiveram como resultado o ganho de peso, perda muscular, questões psicoemocionais, dentre outros. Esse comportamento para os portadores de miopatia foi muito mais danoso, porque os indivíduos tiveram que se afastar do que eu diria ser o único tratamento que temos, que é a fisioterapia e outras terapias.
Em março de 2012 foi publicado no Journal of Child Neurology a “Declaração de Consenso sobre Padrão de Cuidados para Miopatias Congênitas”, em que se recomenda sob certas circunstâncias e cuidados que se faça exercícios aeróbicos regulares, se possível pelo menos duas ou três vezes por semana. Em síntese, pode-se verificar a seguir algumas das diretrizes da referida Declaração:
Exercício Físico: Para manter e maximizar a força muscular, aconselhamos o exercício regular de resistência simétrica, idealmente de caráter concêntrico, e a inclusão de atividades recreativas no plano de exercícios. O exercício deve ser para manter a função muscular. Não há evidências de que o exercício seja prejudicial neste grupo de doenças, como alguns acreditam ser o caso das distrofias musculares. Regimes de exercícios de resistência simétricos devem ser feitos regularmente. Embora não haja evidência direta para especificar a frequência ou intensidade do exercício nas miopatias congênitas, o consenso sugere (como em crianças saudáveis) uma frequência mínima recomendada de 2 a 3 vezes por semana. A fadiga induzida pelo exercício e a dor muscular devem ser evitadas. Se estiverem presentes em um determinado nível de atividade, isso deve ser usado como guia para modular a intensidade do exercício. Esportes de alto impacto devem ser evitados devido ao aumento do risco de lesões.
Ficar em pé: A posição em pé é fortemente recomendada mesmo para crianças extremamente fracas e pode ser facilitada através de estruturas em pé, como maca ortostática, estabilizadores verticais, e órteses. O controle das contraturas ou encurtamentos por meio de programas de fisioterapia de alongamento e órteses ajuda manter a capacidade de ficar em pé. Ficar em pé ajuda no controle da contratura e no desenvolvimento do tronco, da pelve e do controle da cabeça. Ficar em pé também serve como precursor da deambulação e ajuda a promover a autoestima. A resistência e o desenvolvimento ósseo podem ser melhorados quando a pessoa fica em pé. É necessário cuidado ao posicionar os pacientes em estruturas em pé (maca ortostática, estabilizadores verticais, e órteses), especialmente para aqueles com contraturas articulares e osteopenia.
Deambulação e mobilidade assistida: A promoção da mobilidade independente é essencial para pacientes com miopatias congênitas, mas isso também pode ser realizada usando deambulação assistida com a utilização de bengala, ou andador. A utilização de cadeira de rodas também deve ser considerada como importante, pois além de promover a independência, desenvolve a força e o equilíbrio pélvico, do tronco e da coluna vertebral.
Amplitude de movimento articular: As recomendações para manter a amplitude de movimento articular incluem alongamento passivo e ativo-assistido, imobilização estática e progressiva. As órteses são usadas para melhorar o controle postural e minimizar a formação de contraturas. As órteses podem ser usadas para maximizar a mobilidade independente e no contexto de um programa de pé estático. A órtese troncular pode ser usada para estabilizar a coluna nas posições sentada ou em pé. E atenção, a toxina botulínica (Botox) é contra-indicada no músculo esquelético para crianças com doença muscular primária para qualquer indicação. O Botox causa paralisia muscular e quando desaparece seus efeitos em 3 a 6 meses, a atrofia por desuso no músculo pode sobrepor à doença muscular primária, e com o atraso na reabilitação pode fazer com que o paciente tenha mais dificuldade na mobilidade.
UM TESTEMUNHO DE VIDA DE UM PORTADOR DE MIOPATIA CONGENITA CENTRONUCLEAR EM MEIO ÀS ADVERSIDADES DA DOENÇA
A Fundação RYR-1 sediará em julho de 2022 o primeiro Workshop Internacional de Pesquisa em Doenças Relacionadas ao RYR-1, reunindo um grupo internacional de especialistas, cientistas pesquisadores, assim como um seleto grupo de indivíduos afetados por uma doença relacionada ao RYR-1. Neste encontro deverá acontecer um intercambio de informações sobre os mecanismos das doenças relacionadas ao RYR1, a posição atual sobre as pesquisas em curso e perspectivas futuras de tratamentos, mas o mais importante para nós afetados pela doença, é que teremos a oportunidade de expor nossas experiências, queixas, como a doença evolui e nos afeta no dia a dia.
Eu, como portador de Miopatia Congênita Centronulear, fui um dos indivíduos convidados a participar deste encontro, e pretendo na oportunidade oferecer insights pessoais visando ajudar os pesquisadores e clínicos a entender melhor como essa doença afeta nossos corpos nas várias fases e situações da vida. Para melhor conhecimento dos cientistas e pesquisadores presentes, nós convidados, deveremos fazer uma apresentação pessoal, como um histórico de toda a vida sobre nossa relação com a doença.
Em última análise, a partir de nossas histórias, fico na esperança que os cientistas com a compreensão aprimorada, possam mediante as informações recebidas incrementar suas pesquisas para produção de drogas e terapias eficazes em nosso benefício.
Atendendo a sugestão de pessoas que previamente tomaram conhecimentos do meu histórico de vida, reproduzo abaixo o texto base sobre o que será minha exposição no referido evento, o tornando assim de conhecimento de todos.
Meu nome é Orlando, brasileiro, moro em Goiânia, GO, cidade localizada na região centro oeste do Brasil. Minha história de vida começa em 1963, e este texto está focado na minha saúde física. Gostaria de começar dizendo que apesar de não ter sofrido complicações durante a gravidez da minha mãe, nem problemas no parto, meu desenvolvimento físico foi marcado desde o início da minha vida pelo atraso motor e pela hipotonia já notada no primeiro ano de vida. Vale ressaltar que eu já tinha um casal de irmãos fisicamente normais, livres de doenças.
Na época, em meados da década de 1960, diante das limitações científicas e do pouco conhecimento médico disponível em Goiânia, cidade onde morava, aos 7 anos, meus pais decidiram ir em busca de explicações e diagnóstico sobre o que estava me afetando fisicamente, e assim fomos para São Paulo, um grande centro médico do Brasil. Eu já apresentava marcos motores preocupantes, como a manobra de Gowers, dificuldade para subir degraus, dentre outros. Uma biópsia muscular veio comprovar relatando sinais inusitados e inespecíficos no tecido (somente coloração de H&E e trinômio de Masson), mas o neurologista limitou-se a dizer que eu estava acometido por uma potencial doença neuromuscular, e não foi indicado nenhum tratamento a ser realizado.
Em 1970, nasceu minha irmã, o que na época causou à família um misto de alegria pelo seu nascimento, mas preocupação porque ela já dava sinais claros de que também era acometida pela mesma doença inespecífica que me acometia. Meus pais, preocupados com a situação decidiram levar eu e minha irmã, ainda bebê, para outro grande centro médico, desta vez para o Departamento de Estudos Neurológicos da Universidade Federal do Rio de Janeiro. Lá, mais exames foram realizados, incluindo um eletromiografia, e desta vez nos foi dada uma hipótese de diagnóstico clínico de um tipo de Distrofia Muscular. Na ocasião o médico conversou com meus pais sobre as características e prognóstico daquela doença, assim como recomendou cuidados especiais com exercícios físicos para evitar uma possível progressão da doença, mas nada foi indicado como tratamento.
Durante minha infância e adolescência, mesmo considerando minhas dificuldades e as claras diferenças em relação às outras crianças, fui muito ativo, e tentei fazer tudo, nadei, andei de bicicleta, enfim, brinquei muito. Fui criado e ensinado de uma forma que não focava no que eu não podia fazer, mas no que eu queria e era capaz de fazer dentro das minhas habilidades. Ao longo dos anos, experimentei um padrão de piora física lenta, mas constante, tais como a dificuldade em subir escadas, levantar de uma cadeira, andar, manter o equilíbrio e riscos de queda. Emocionalmente também sofri muito por vezes me sentir diferente dos meus colegas, mas ao mesmo tempo em que fui fortemente apoiado pelos amigos e familiares, me fazendo sentir normal, mesmo sendo uma criança fisicamente anormal.
As dificuldades que sempre enfrentei na vida, me fizeram desenvolver um instinto de superação e de busca pela Independência. Casei-me muito jovem, aos 19 anos, quando ainda cursava faculdade. Já casado, aos 21 anos, mudamos para os EUA fazer meu MBA. Naquela época pensavaos em ter filhos, mas diante da falta de conhecimento e certeza sobre a doença que me acometia, por termos ouvido falar do MDA - Associação de Distrofia Muscular, fomos lá em busca de respostas e aconselhamento. Fomos a uma clínica associada em Austin, Texas, cidade onde morávamos, e consultados pelo Dr Jerry Tindel, que após uma bateria de exames, recebi o diagnóstico de Distrofia Muscular, sugerindo ser do tipo FSH-Facioscapulohumeral. Na consulta me foi explicado sobre a gravidade da doença, seu prognóstico, a probabilidade de ter um filho também afetado pela doença, e concluir dizendo que em poucos meses eu estaria em cadeira de rodas, contudo, nenhum tratamento me foi prescrito.
De volta ao Brasil, a vida continuou, e nos anos que se seguiram, as décadas de 1980 e 1990, minhas dificuldades físicas aumentaram, era sinal da evolução da miopatia, eu não conseguia mais me levantar de uma cadeira, e precisava usar uma bengala para apoiar minha caminhada e equilíbrio, mas ao contrário do que o médico do MDA tinha dito, eu não precisava de usar cadeira de rodas. Neste período estive muito envolvido com meus projetos profissionais e familiares. Eu tive um casal de filhos, e ambos nunca mostraram sinais de serem afetados pela doença. Luciano, meu filho mais velho, apesar de ter falecido aos 23 anos vítima de leucemia, durante sua vida como prova de sua capacidade física se tornou atleta de triathlon, e conquistou a medalha de bronze aos 20 anos no Campeonato Mundial de Triatlo em Vancouver, Canadá, o que demonstrou não ser portador de nenhuma miopatia. Priscila, nossa filha caçula, hoje é médica e casada, e é fisicamente normal, mas carrega uma mutação no gene RYR-1.
No ano de 2000, incomodado com a progressão e piora física com aumento da limitações impostas pela doença, porém não tão grave como dizia em 1985 o médico do MDA, fui novamente em busca de respostas, e desta vez acreditando na evolução científica marcada pela mudança do século. Procurei pelo que existia de mais avançado na ciência, e fui consultado pela Dra Mayana Zatz, geneticista do Departamento de Genética Humana da USP - Universidade de São Paulo, em busca de um diagnóstico, contudo, mais uma vez me viram como portador de uma Distrofia Muscular, mas novamente sem um laudo conclusivo.
Inconformado com a imprecisão dos hipotéticos diagnósticos que recebi no decorrer da minha vida, finalmente, em 2007, aos 44 anos, conheci o Dr Acary Bulle Oliveira, neurologista e chefe do Setor de Doenças Neuromusculares da UNIFESP - Universidade Federal de São Paulo, e sendo consultado por ele, finalmente recebi um diagnóstico clínico e conclusivo, corroborado por biópsia muscular que evidenciou anormalidades da rede intermiofibrilar típicas de uma Miopatia Congênita Centronuclear, bem como resultado de exame genético e pesquisa com sequenciamento total do exoma mostrando alteração (mutação) no gene RYR1.
Dada a raridade da miopatia que me afeta, minha idade e situação física, e evolução da doença, em 2017 fui convidado a visitar o NIH - National Institutes of Health, Bethesda, Maryland, EUA, e consultado pelo Dr Carsten Bonnemann, MD., Senior Investigator and Chief of the Neuromuscular and Neurogenetic Disorder me coloquei a disposição para participar de eventual estudo de pesquisa. Esse momento foi muito impactante na minha vida, pois pela primeira vez percebi que haviam pessoas interessadas na minha doença, e que já estavam desenvolvendo pesquisas em busca de tratamento e até de cura. Na época, outro grande marco foi ter sido apresentado à Fundação RYR1, que é uma associação que visa reunir pessoas afetadas por doenças relacionadas ao RYR1 de todo o mundo, promover a troca de experiências entre os participantes (afetados e familiares), além de apoiar cientistas e laboratórios que trabalham no desenvolvimento de medicamentos para tratamento, a até a terapia genética curativa. Enfim, toda essa experiência me fez ver e prever algo que nunca tinha pensado, que é a cura, ou pelo menos a existência de um medicamento para tratar ou aliviar os efeitos da Miopatia Congênita Centronuclear.
(este texto foi escrito originalmente em inglês, e traduzido para o português para esta postagem)
EXISTE EM TODO MUNDO UM DIA ESPECIALMENTE DEDICADO PARA CHAMAR A ATENÇÃO DA SOCIEDADE SOBRE AS DOENÇAS RARAS
O Dia das Doenças Raras acontece anualmente em todo o mundo, geralmente no último dia de fevereiro. Essa data foi criada com vistas no impacto causado na vida dos indivíduos afetados por uma destas doenças. Ela tem como objetivo aumentar a conscientização dos formuladores de políticas, público em geral, assim como estimular os desafios em pesquisas científicas de tratamentos sobre as doenças raras.
Doença Rara, segundo a Organização Mundial de Saúde (OMS), é a doença que afeta até 65 pessoas em cada 100 mil indivíduos, ou seja, 1,3 para cada 2 mil pessoas. Nos E.U.A. é considerado uma Doença Rara se ela afetar menos que 200.000 indivíduos.
Cerca de 8.000 doenças raras afetam pessoas por todo mundo. Essas doenças são caracterizadas por uma ampla diversidade de sinais e sintomas e variam não só de doença para doença, mas também de pessoa para pessoa acometida pela mesma condição. Por volta de 80% destas doenças têm origem genética, são crônicas, progressivas, degenerativas e muitas vezes com risco de morte e na maioria dos casos não existe uma cura eficaz existente. Como as doenças raras geralmente são difíceis de diagnosticar, pode levar anos para obter um diagnóstico preciso, e mesmo assim, na maioria das vezes, sem ter um tratamento disponível, porque menos de 5% das doenças raras têm um tratamento aprovado pelos órgãos oficiais.
As doenças raras, quando beneficiadas por um tratamento via droga, é feito pela prescrição de um chamado “medicamento órfão”. Este termo é um status dado por um órgão regulatório de saúde oficial de cada país a medicamentos que tratam doenças raras. Para o FDA, órgão americano, é considerado medicamento órfão aqueles que tratam doenças que atingem menos de 200.000 indivíduos. Para a União Europeia, estes medicamentos são aqueles que tratam não mais do que cinco em cada 10 000 pessoas e as doenças tratadas com eles sejam crônicas debilitantes ou coloquem a vida em risco. De acordo com a Anvisa, órgão brasileiro, os medicamentos órfãos são indicados para incidência de doenças menor que 5 em 10 000 indivíduos.
Com relação à Miopatia Congênita Centronuclear, tema central deste site, doença muscular causada por uma mutação genética no RYR-1, por sua pequena incidência na população, esta ainda tem sua taxa de incidência desconhecida.
Por fim, gostaria de destacar que mesmo sendo afetado por uma doença que levou 44 anos para ser diagnosticada, classificada como “Doença Rara”, e fazendo parte de um grupo em que as estatísticas nos fazem invisíveis à sociedade, vivo atualmente um momento em que me sinto totalmente acolhido. Desde que conheci o NIH - National Institute of Health, e a Fundação RYR1, através deles descobri que existem médicos, cientistas e laboratórios que se preocupam e trabalham em busca de tratamento e cura para as doenças relacionadas ao RYR1.
Abordagem Respiratória na Miopatia Centronuclear
Por: Alessandra C. Dorça
As Miopatias Congênitas Centronucleares (CNMs) são um grupo de doenças neuromusculares hereditárias classificadas como miopatias congênitas. Embora vários genes causadores tenham sido identificados, alguns pacientes não abrigam nenhuma das mutações atualmente conhecidas. Esses diversos distúrbios têm características histológicas comuns, que incluem uma alta proporção de fibras musculares com núcleo central, e atributos clínicos de fraqueza muscular e insuficiência respiratória.
Neste texto será abordado especificamente sobre questões respiratórias. As alterações respiratórias podem se manifestar inicialmente durante o sono, mas sintomas diurnos e dificuldade de proteção da via aérea predominam à medida que a disfunção muscular respiratória evolui para mais grave.
A avaliação respiratória do indivíduo afetado é fundamental para acompanhar o comprometimento muscular. A capacidade muscular respiratória pode ser avaliada usando uma variedade de testes clínicos, como análise de capacidades pulmonares, forças musculares inspiratórias e expiratórias, e pico de fluxo de tosse.
O comprometimento respiratório costuma ser a maior causa de morte, já que algumas formas mais graves apresentam hipotonia e fraqueza de região faríngea, ocasionando distúrbios de deglutição e comprometimento respiratório recorrente devido a pneumonias bronco aspirativa. Nestes casos a indicação precoce da sonda gástrica pode ser de fundamental importância.
A disfunção dos músculos respiratórios pode ser agravada por restrições mecânicas que aumentam a carga respiratória, incluindo baixa complacência pulmonar, defeitos restritivos musculoesqueléticos na parede torácica e cifoescoliose. Com a evolução da doença, a fraqueza da musculatura bulbar ocasiona episódios recorrentes de engasgos, e a troca espontânea da consistência alimentar pode facilitar a mastigação favorece a deglutição. As manifestações funcionais da insuficiência dos músculos respiratórios podem incluir distúrbios do sono, fadiga, tosse ineficaz e hipoventilação.
As várias mutações genéticas das miopatias possuem progressão diferente, mas a diminuição da capacidade motora ocasiona, por consequência a diminuição do volume corrente e do tempo inspiratório com aumento concomitante da carga elástica e diminuição comprimento-tensão musculares em volumes pulmonares mais altos. O resultado funcional é um padrão respiratório rápido e superficial, necessidade de suporte ventilatório para manter a capacidade elástica muscular, e evitar a restrição severa da caixa torácica.
O quadro a seguir apresenta as características clínicas nas miopatias causadas por mutações nos diversos genes:
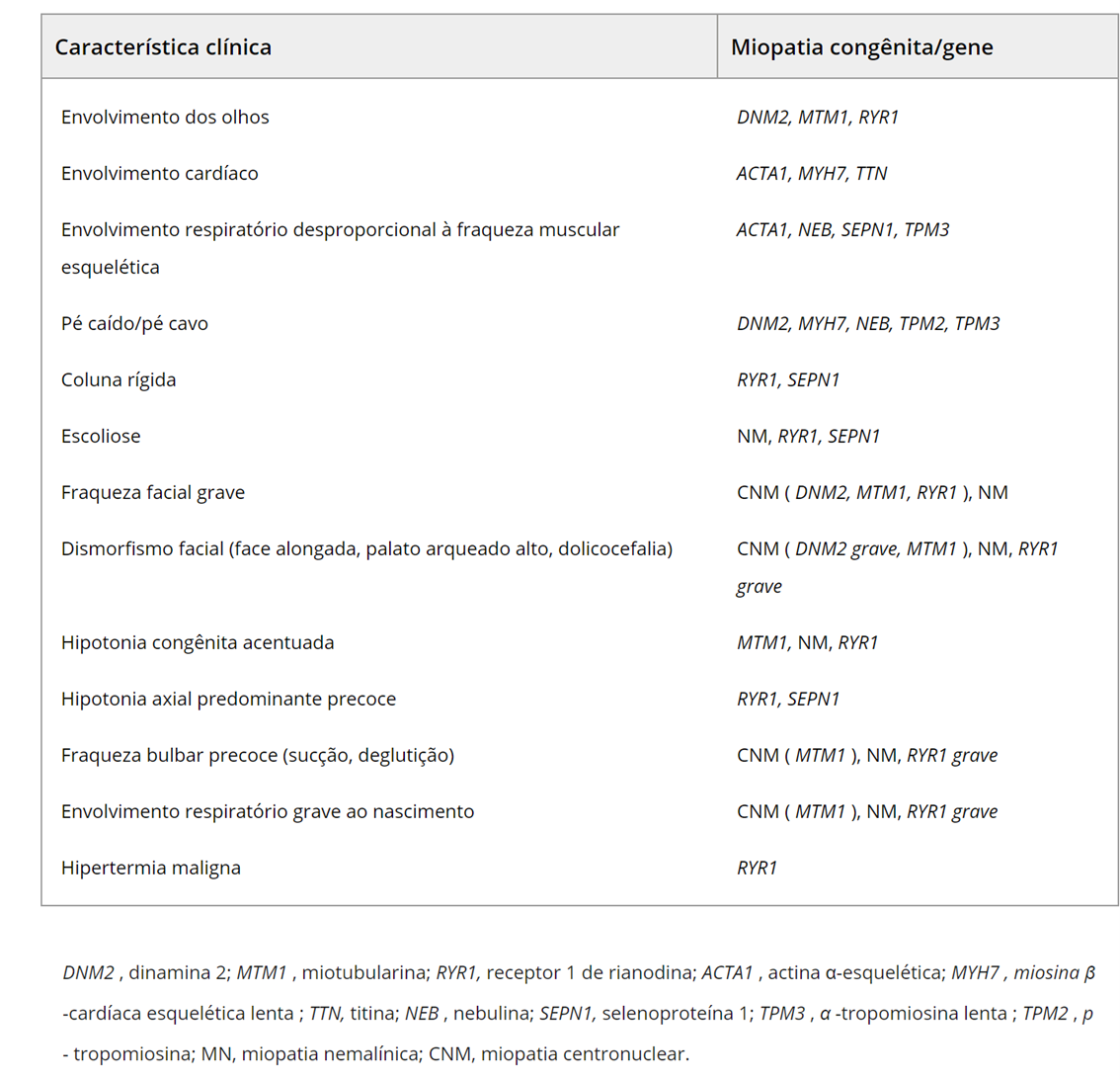
Cuidados Respiratórios nas Miopatias Congênitas Centronuclear (CNMs)
As várias mutações genéticas que causam as miopatias centronucleares apresentam progressões variadas, principalmente relativo à velocidade da evolução da doença. No caso, a progressão lenta ou mais acelerada traz a necessidade de cuidado respiratório próximo, fazendo assim necessário uma Avaliação Respiratória Funcional, visando uma objetiva e eficaz Abordagem Terapéutica Respiratória (tratamento).
Avaliação Respiratória Funcional
A avaliação respiratória funcional é capaz de acompanhar a velocidade de progressão da doença, e assim direcionar as abordagens necessárias em cada momento da doença, e deve ser realizada a cada 6 meses, desta forma as abordagens preventivas poderão minimizar as complicações da patologia, como segue:
1 - Avaliação da capacidade pulmonar
CVF (Capacidade Vital Forçada) - A prova de função pulmonar é capaz de predizer a capacidade vital e correlacionar com a idade e o peso, desta forma é possível verificar o comprometimento no volume pulmonar devido a diminuição da força dos músculos respiratórios. A CVF (Capacidade Vital Forçada) orienta a necessidade de suporte respiratório.
2 - As forças musculares respiratórias, PIMAX (pressão inspiratória máxima), e PEMAX (pressão expiratória máxima), são mensuradas através da Manovacuometria.
PIMAX - é a pressão inspiratória máxima, e reflete o valor da força muscular inspiratória, ou seja, a força diafragmática. O diafragma é o maior musculo da respiração e corresponde a 50 % da capacidade de geração de volume pulmonar. A fraqueza diafragmática favorece a hipoventilação noturna, a dificuldade de geração de volume pulmonar e restrição respiratória.
PEMAX - é a pressão expiratória máxima, e reflete o valor da força da musculatura abdominal, que é responsável pela centralização da força do tronco, auxilia a tosse e favorece o movimento de expiração e auxilia explosão da tosse, o que proporciona a melhor proteção pulmonar
3 - Pico de Fluxo de Tosse (PFT)
A tosse é o principal mecanismo de proteção da via aérea. Para a efetividade da tosse é necessário a sinergia de vários músculos da região laríngea, glote, e músculos abdominais. A avaliação do pico de fluxo de tosse é capaz de predizer a força desta musculatura, e a sincronia no movimento. O comprometimento da musculatura bulbar tem como resposta inicial a diminuição do pico de tosse.
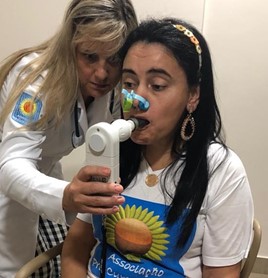
Avaliação respiratória das forças musculares ( Micro RPM), prova de função pulmonar (Mini Spir)
Abordagem Terapáutica Respiratória - Tratamento
A fisioterapia respiratória é responsável pela minimização dos comprometimentos respiratórios nas miopatias centronucleares, pois interfere na progressão da perda muscular, minimiza as complicações da perda de capacidade pulmonar, mantem a funcionalidade respiratória como fala, deglutição e tosse. Fazem parte das estratégias utilizadas no tratamento:
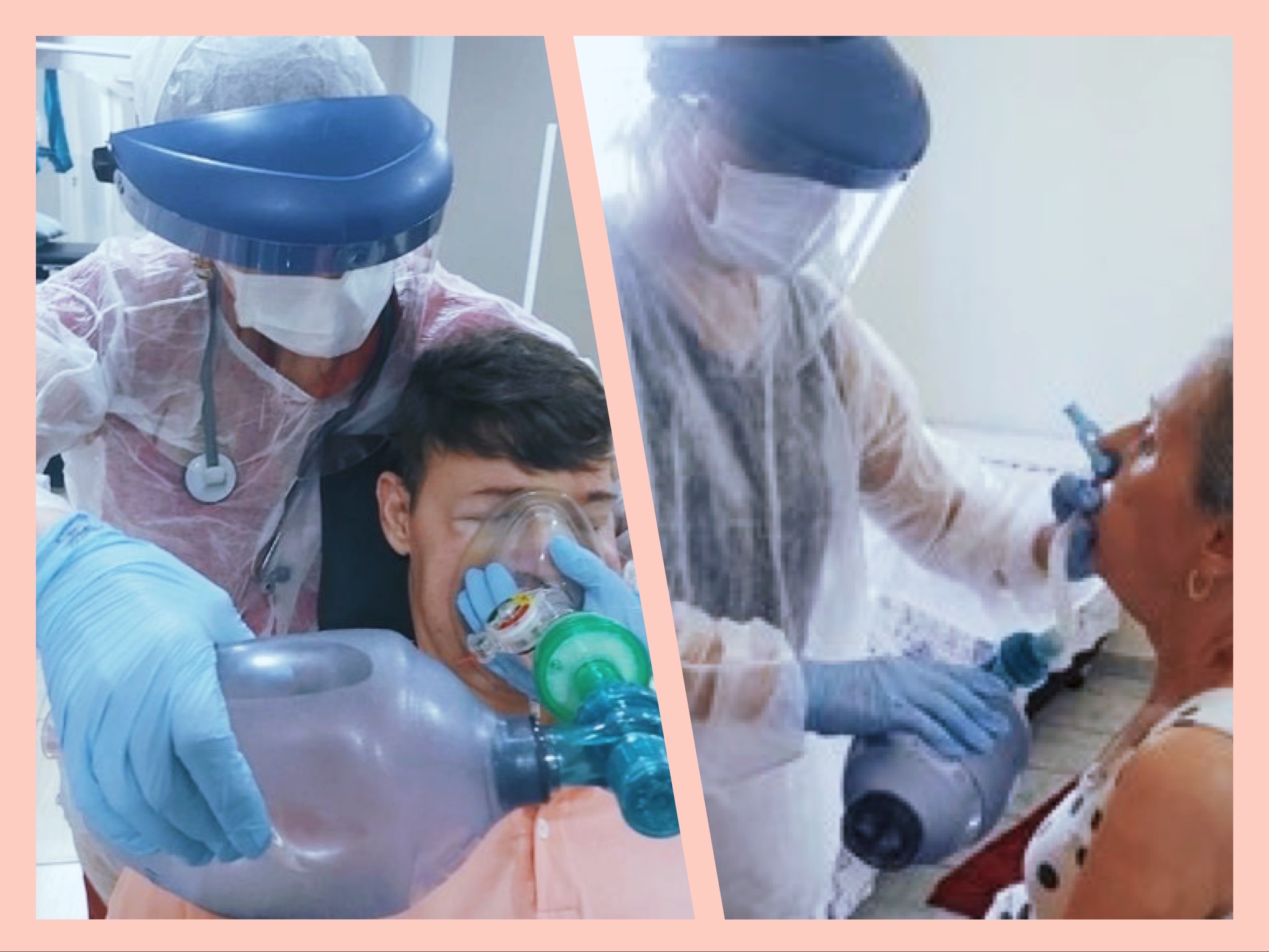
Fig 1 -Exercício de empilhamento de ar \ Fig 2 - Exercícios respiratórios para manutenção da capacidade pulmonar e treinamento da musculatura bulbar (ambos utilizando a bolsa de insuflação (ambu)
O cuidado respiratório deve ser uma prioridade nos portadores de Miopatia Centronucelar, e o profissional deve ser capaz de identificar a alteração funcional e correlacionar com a melhor terapia a ser aplicada, sendo esta a melhor estratégia para evitar maiores complicações para o paciente.
Bibliografia Consultada:
Smith BK, Goddard M, Childers MK. Avaliação respiratória nas miopatias centronucleares. Nervo Muscular 2014;50(3):315-326. doi:10.1002/mus.24249
Benditt JO. Respiratory Care of Patients With Neuromuscular Disease. Respir Care. 2019 Jun;64(6):679-688. doi: 10.4187/respcare.06827. PMID: 31110036.
OS PORTADORES DE DOENÇAS RELACIONADAS AO RYR-1 E SEUS FAMILIARES NÃO ESTÃO SÓS E SUAS CAMINHADAS
No ano de 2018, enquanto participava como convidado de uma pesquisa conduzida pelo Dr. Carsten Bonnemann, no NIH - National Institutes of Health, em Bethesda, MD, E.U.A., em meio a tantas informações que estava obtendo, tive a grata satisfação de tomar conhecimento da existência da Fundação RYR-1, sediada em Pittsburgh, PA. Ansioso e eufórico com tudo que estava vivendo naqueles dias, no momento imediato fiz contato com a organização para conhecer e saber mais sobre seus propósitos, e o resumo de tudo aquilo se traduziu em um grande marco na minha vida, pois descobri depois de muito tempo que eu não vivia solitário uma doença desconhecida, e sem ninguém se importar com o que sentia, e o quê poderia ser feito sobre tudo aquilo.
Assim, é com muito respeito e gratidão que gostaria de apresentar a Fundação RYR-1. A Fundação RYR-1 foi iniciada por membros da Família Goldberg, que foram afetados por uma doença relacionada ao RYR-1. Atualmente, não há nenhuma outra organização que exista apenas para defender e atender às necessidades dos pacientes com doenças relacionadas ao RYR-1. O objetivo da Fundação é preencher esse vazio tão necessário.

A missão da Fundação RYR-1 é apoiar pesquisas que conduzam a um tratamento eficaz ou cura para doenças relacionadas ao RYR-1.
Para cumprir esta missão, a Fundação tem vários objetivos:
A Fundação RYR-1 é liderada por uma equipe dedicada de indivíduos. Isso inclui o Conselho de Administração, o Conselho Consultivo Científico, e o Conselho de Consultores, e as operações diárias são lideradas pelo Diretor do Programa.
Contudo, ao conhecer a Fundação RYR-1, o que mais me impressionou com esta organização foi, acima das questões científicas, pela personalidade que seus trabalhos são conduzidos, e tudo isso tendo à sua frente o Dr Michael F. Goldberg, MD, MPH, que também é portador de uma miopatia relacionada ao RYR-1, o que lhe confere uma grande propriedade por entender todas as questões ligadas à doença, e assim gerir a organização. Gostaria de compartilhar uma mensagem escrita pelo Dr Michael, transcrita do site da fundação (@RYR1.org):
“Embora raras, as doenças relacionadas ao RYR-1 são a forma mais comum de “miopatia congênita” - doença muscular causada por uma mutação genética. As manifestações mais comuns incluem: fraqueza muscular leve a moderada (por exemplo, dificuldade em subir escadas), fraqueza grave (por exemplo, necessidade de assistência de cadeira de rodas e suporte respiratório), cãibras e dores musculares e uma reação potencialmente fatal à anestesia, chamada hipertermia maligna. No entanto, apesar da prevalência desta doença e da gravidade desses sintomas, a Fundação RYR-1 é a única organização que existe apenas para apoiar a pesquisa sobre esta doença. Além disso, a Fundação RYR-1 atende às necessidades e defende indivíduos e famílias afetadas por doenças relacionadas ao RYR-1.
Em um período muito curto de tempo, a Fundação RYR-1 tornou-se líder no campo de doenças relacionadas ao RYR-1.
Devido ao apoio de nossos muitos benfeitores generosos, o trabalho da Fundação levou a avanços importantes no conhecimento científico. Além disso, formamos uma comunidade mundial de indivíduos afetados que podem encontrar conforto e apoio sabendo que não estão sozinhos e que agora nossa organização existe para ajudá-los.
A necessidade de tratamentos ou cura é urgente. Com o seu apoio, a Fundação RYR-1 pode continuar a financiar cientistas de renome mundial, expandir a comunidade RYR-1 e tornar nosso slogan, “Strength in Numbers,” (“Força em Números”), uma realidade.”
- texto/mensagem transcrita da brochura promocional da Fundação RYR-1 -
Conheça e tenha maiores informações sobre a Fundação RYR-1 através do link : https://www.ryr1.org/
No curso de nossas vidas temos a todo tempo que fazer escolhas. Durante mais de 40 anos vivi uma condição física que me limitava muito fisicamente, que se agravava com o passar do tempo, e que me impunha a todo tempo diante do questionamento sobre qual doença era aquela que me afetava. Sofri com dor física, ansiedade, sentimento de rejeição, discriminação, incapacidade, enfim, mas como disse Stephen Hawking, “a vida seria trágica, se não fosse engraçada”, e talvez por ter aprendido os princípios da resiliência, superação, e de ser grato a Deus por tudo que sou e tenho, “SORRIR” foi a grande escolha que fiz, e isto serviu como antídoto para enfrentar todas as minhas dificuldades. Pode parecer irônico, mas quando digo “SORRIR”, me refiro a levar a vida com leveza, focando no positivo, desprezando as impossibilidades e limitações que me são impostas, além de exercitar e potencializar as possibilidades que tenho, superando assim meus limites.
Aconteceu que com os avanços da medicina e da genética, finalmente aos 44 anos obtive um diagnóstico de uma doença rara, doença esta que afeta o gene RYR-1, o qual é responsável pelo funcionamento de todos os músculos esqueléticos, que tem o padrão de progressividade no seu agravamento, e que se chama Miopatia Congênita Centronuclear, …e daí perguntei a mim mesmo: o que mudou na minha vida ? …nada ! …só acharam o nome e endereço para o quê sempre me afetou, e decidi que nada mudaria, e que seguiria minha vida da mesma forma, porque já tinha feito lá atrás a opção de SORRIR para vida, mas só que a partir daquele momento, com uma analogia ao RYR-1, iria SORRYR-1.

Apesar de que durante toda minha história de vida, muitas coisas podem ter me incomodado, fosse fisicamente, ou emocionalmente, a falta informação, e conhecimento sobre a doença que me afetava, essa sim, me chateou muito. Acredito que o conhecimento ou tratamento de qualquer doença tem o princípio com a informação, e a informação não se limita a quem trata, mas também a quem é tratado. E como disse o escritor Aldous Huxley, “Conhecimento não é aquilo que você sabe, mas o que você faz com aquilo que se sabe”, portanto, não adianta ter o conhecimento e não o utilizar para algum propósito.
Por saber sobre a dificuldade no acesso às informações gerais sobre a Miopatia Congênita Centronuclear, e pela vivência como portador da doença, foi que decidi criar este blog, visando a informação do conhecimento, mas que também possa ser um canal de interação através da troca de experiências pessoais e científicas, contribuindo positivamente de alguma forma com aqueles que estão inseridos no contexto das doenças relacionadas ao RYR-1, seja como indivíduo afetado, familiar, pessoas de seu convívio, ou profissional de saúde, ou enfim, a todo aquele que possa se interessar sobre o assunto.
O DIAGNÓSTICO DE UMA DOENÇA RELACIONADA AO RYR-1 PODE TER SIDO UMA TAREFA DIFÍCIL NO PASSADO, MAS NÃO ATUALMENTE
As doenças relacionadas ao RYR1, são denominadas como “doenças raras” pela Organização Mundial da Saúde (OMS) segundo critérios de incidência, raridade, gravidade, e diversidade.
As doenças relacionadas ao RYR1 fazem parte de um grupo de doenças genéticas em que os músculos não funcionam adequadamente, também chamadas de miopatias relacionadas ao RYR1. Em geral, pessoas com doenças relacionadas ao RYR1 têm fraqueza muscular ou tônus muscular pobre. Em alguns casos, crianças podem demorar mais para sentar, engatinhar e andar. Os indivíduos portadores destas miopatias também podem ter problemas na coluna vertebral, músculos oculares, mastigação, deglutição e respiração. Além disso, essas miopatias podem causar uma ampla gama de sintomas de fraqueza leve a grave, às vezes exigindo assistência de cadeira de rodas e suporte respiratório, a uma potencial reação fatal a certas formas de anestesia conhecidas como Hipertermia Maligna (MH), e certas formas podem levar à intolerância ao calor, insolação por esforço, e uma forma grave de colapso muscular, chamada rabdomiólise.
Devido ainda ao pouco conhecimento médico-científico das doenças relacionadas ao RYR1, alguns dos sintomas das doenças relacionadas ao RYR1, por ser semelhantes com outras desordens, podem dificultar o processo de identificação da patologia, pois algumas condições passam despercebidas por médicos generalistas, o que muitas vezes gera um subdiagnóstico, portanto, é quando em busca de um diagnóstico preciso e específico para a identificação da doença, é essencial encontrar um centro clínico e um profissional especialista em doenças neuromusculares.
Pela dificuldade de diagnóstico e mesmo na distinção entre os vários tipos de doenças musculares relacionadas ao RYR1, é importante observar as seguintes características físicas, sinais e sintomas normalmente presentes no portador de uma das doenças...a conferir a seguir:
Contudo, para um laudo conclusivo no diagnóstico da doença relacionada ao RYR1, os médicos podem solicitar os seguintes exames:

Fonte das Figuras: livescience.com, theindepthgenealogist.com,
Posição e Recomendações da “World Muscle Society” (WMS) Versão 2 – atualização em 06/04/2020 - Brazil
Na categoria de doenças neuromusculares (DNM) se enquadra um grupo amplo de diferentes diagnósticos com níveis variados de disfunção, mesmo entre pessoas com o mesmo diagnóstico de doença específica. Portanto, torna-se difícil fazer recomendações que se apliquem em geral. As que se seguem, são recomendações para um numeroso grupo dentre estas condições. Se destinam primariamente a pacientes, cuidadores, neurologistas e médicos generalistas. Tem ainda a intenção de informar especialistas em doenças neuromusculares, com respeito às necessidades básicas requeridas, endereçando os questionamentos frequentes. Links detalhados de referência foram incluídos
Nota da tradução: Este documento deve ser aplicado às características e especificidades de atendimento em cada país. Pequenos ajustes foram feitos, para a população Brasileira.
1. As pessoas com DNM estão em maior risco?
Associações neurológicas nacionais e redes neuromusculares (Associação Britânica de Neurologistas, Rede Europeia de referência EURO-NMD, outras) produziram um guia no impacto do Covid-19 para doenças neurológicas e o seu manejo. Estes documentos definem o risco de evolução grave para Cocid- 19 como alto ou moderado em todas exceto as formas mais brandas de DNM. Características que conferem riscos altos ou muito altos de acometimento mais grave incluem, por exemplo:
2. O que pessoas com DNM podem fazer para evitar a infecção?
Covid-19 se dissemina por gotículas, quando uma pessoa infectada tosse, espirra ou fala, ou ainda toca com sua mão com estas gotículas infectadas em diferentes superfícies (maçanetas, interruptores e outros diferentes objetos de uso diário). Pessoas com DNM e as demais devem seguir as seguintes precauções:
3. Que consequências a infecção pelo Covid-19 pode trazer para os tratamentos das pessoas com DNM?
4. O que precisa ser feito para assegurar suporte ventilatório no isolamento.
5. Quando pacientes com DNM devem buscar unidade hospitalar?
Os pacientes com DNM são considerados grupo de risco para COVID-19. A internação hospitalar deve ser evitada, se possível, mas não deve ser adiada quando necessário. Esta pode ser uma decisão difícil. Pessoas com NMD precisam estar cientes de que:
6. Tratamentos para Covid-19 podem afetar as DNM?
7. O que os especialistas em DNM deveriam fazer para dar suporte aos médicos de emergência e de terapia intensiva no cuidado de pacientes com DNM ?
As decisões sobre a admissão de doentes em unidades de cuidados intensivos podem ser afetadas por problemas previsíveis ou existentes de capacidade. Pode ter sido instituído um sistema de triagem e isso pode ter consequências práticas e éticas.
8. Qual o suporte os centros de acompanhamento de pacientes com DNM deveriam providenciar?
Novas informações e resultados sobre o Covid-19 relevantes para a doença neuromuscular:
Problemas cardíacos:
Tratamento imunossupressor em pacientes com doença neuromuscular:
Outras informações:
Os documentos estão disponíveis em:
Informações em sites locais:
Referências:
Autores do documento:
Elaborado por:
Documento endossado por:
Tradução e adaptação para o Brasil
Depto de Neuropediatria, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ
Depto. de Neurologia, Faculdade de Medicina (FMUSP), Universidade de São Paulo, São Paulo, SP
Depo. de Pediatria, Faculdade de Medicina, Universidade Federal de Minas Gerais, Belo Horizonte, MG
Centro de Estudos do Genoma Humano e Células Tronco, IB Universidade de São Paulo, São Paulo, SP

A Fundação RYR-1 é uma organização que existe para defender e atender às necessidades dos pacientes com doenças relacionadas ao RYR-1, e visa apoiar pesquisas que conduzam a um tratamento eficaz ou à cura de doenças relacionadas ao RYR-1, a até promover o relacionamnto pessoal de indivíduos afetados, seus familiares, cientistas, através de conferências de âmbito internacional.
ZNM-ZUSAMMENSTARK.ORG
ZNM – Zusammen Stark! e.V. - Associação organizada com esforços próprios em apoio aos afetados com miopatia miotubular e outras miopatias centronucleares (CNM = ZNM), e representada pessoas de famílias com CNM na Alemanha, Holanda e Áustria. A organização tem como principal objetivo conectar as pessoas afetadas e suas famílias entre si, assim como trocar experiências e apoiar uns aos outros, dando voz às famílias, compartilhar informações e pesquisas mais recentes sobre CNM, patrocinador bolsas para pesquisas sobre uma cura, e networking com outras organizações.

C-MyPath - Grenoble Institut des Neurosciences
O C-MyPath (Cellular Myology and Pathology) foi criado em 2007 pela INSERM e University Joseph Fourier, como sendo um centro de pesquisas do Grenoble Institute of Neuroscience, na França, e é composta por cientistas, geneticistas, e médicos que dedicam seus trabalhos ao entendimento e pesquisas das doenças neuromusculares.

Malignant Hyperthermia Association of the United States
A Associação de Hipertermia Maligna dos Estados Unidos (MHAUS) é uma organização sem fins lucrativos A missão da MHAUS é promover informações sobre cuidados ideais e compreensão científica sobre a Hipertermia Maligna e doenças relacionadas. MHAUS tem quatro objetivos principais: 1. Educar todo o espectro de profissionais de saúde para que a Hipertermia Maligna seja rapidamente reconhecida e tratada em tempo e adequadamente por pessoas em todas as disciplinas médicas; 2. Aconselhar os profissionais envolvidos e preparar todas as instalações médicas nos Estados Unidos para diagnóstico e tratamento imediato no caso de um episódio de Hipertermia Maligna; 3. Ajudar os indivíduos suscetíveis à Hipertermia Maligna (HM) e as suas famílias a aprenderem a viver com o risco ou suscetibilidade à HM, assim como partilhar com eles a experiência e o conhecimento acumulados sobre a HM; 4. Incentivar e apoiar a pesquisas sobre Hipertermia Maligna, especialmente sobre um teste de diagnóstico não invasivo de alta precisão. Graças aos esforços da MHAUS e de organizações semelhantes em todo o mundo, a taxa de mortalidade foi marcadamente reduzida. Além disso, estes esforços ajudaram a identificar formas de Hipertermia Maligna (HM) que não eram anteriormente reconhecidas pela comunidade médica.

